
Progress in controllable asymmetric coordination studies of single-atom catalysts in energy catalysis
-
摘要:
能源已成为人类社会发展的重要基础,在国民经济中占有举足轻重的地位. 单原子催化剂(SACs)已成为一种很有前途的材料,并且在化学和能源领域都有广泛的应用. 因此,开发具有高活性、高稳定性和低成本的SACs具有重要的实际意义. 特别是SACs中的不对称配位结构,在催化性能上超越了传统的M—N4活性位点,引起了人们的迅速关注. 在氧还原反应(ORR)这一燃料电池技术的关键步骤中,具有不对称配位结构的SACs通过优化活性位点的电子性质来提高催化效率. 这将导致活化能的大幅降低,从而提高电流密度和能量转换效率,从而加速燃料电池的商业化. 同样,在CO2还原反应(CO2RR)中,这些催化剂可以精细地调节CO2分子的吸附和活化,促进CO2选择性和有效地转化为有价值的化学物质,如甲醇和一氧化碳. 这种能力为碳回收技术提供了巨大的潜力. 此外,不对称SACs在解决环境挑战方面显示出显着的希望,特别是在硝酸盐还原反应(NO3RR)中,它们有效地将有害硝酸盐转化为惰性氮,从而有助于环境保护和水质改善. 本文总结了几种典型的非对称单原子催化剂结构,即M—N4—Y(Y为轴向杂原子),M—Nx—Y(Y为非金属原子),M—Nx和M—M结构. 系统介绍了这些先进催化剂的可控合成及其在氧还原反应(ORR)、CO2还原反应(CO2RR)和硝酸根还原反应(NO3RR)等电催化反应中的研究进展和应用. 最后,对目前SACs的挑战和未来的发展前景进行了展望.
Abstract:Energy is foundational for the advancement of human society, occupying a pivotal role in the national economy. Single-atom catalysts (SACs) are a promising catalyst material in the chemistry and energy fields owing to their high activity, high efficiency, adjustable electronic structure, and low price. Therefore, the development of SACs with high activity, superior stability, and low cost holds significant practical importance. In particular, asymmetric coordination structures in SACs have gained attention over traditional M—N4 active sites in terms of catalytic performance. In the context of the oxygen reduction reaction (ORR), a key step in fuel cell technology, SACs with asymmetric coordination structures have demonstrated enhanced catalytic efficiency by optimizing the electronic properties of active sites. This leads to a substantial reduction in activation energy, resulting in improved current densities and energy conversion efficiencies, thus accelerating the commercialization of fuel cells. Similarly, in the CO2 reduction reaction (CO2RR), these catalysts can fine-tune the adsorption and activation of CO2 molecules, promoting the selective and efficient conversion of CO2 into valuable chemicals such as methanol and carbon monoxide. This capability offers significant potential for carbon recycling technologies. Moreover, asymmetric SACs have shown remarkable promise in addressing environmental challenges, particularly in the nitrate reduction reaction (NO3RR) by efficiently converting harmful nitrates into inert nitrogen, which contributes to environmental protection and water quality improvement. In general, axial coordination in electrocatalysts enhances electrocatalytic reactions such as CO2RR and ORR by fine-tuning the electronic structure of metal centers to optimize reaction kinetics and stabilize the catalyst. This coordination facilitates the efficient desorption of catalytic intermediates while mitigating side reactions, leading to improved catalyst durability and enhanced electrochemical stability. This review provides a comprehensive overview of several typical asymmetric SAC structures, including M—N4—Y (where Y represents an axial heteroatom), M—Nx—Y (where Y is a nonmetal atom), M—Nx, and M—M configurations. We systematically review the controlled synthesis of these advanced catalysts, highlighting their recent progress and applications in electrocatalytic reactions such as ORR, CO2RR, and NO3RR. Finally, the challenges and future prospects of SACs in terms of synthesis, performance, and underlying mechanisms are critically discussed. While SACs have made significant progress, issues such as the precise control of atomically dispersed sites, stability under reaction conditions, and understanding the detailed catalytic pathways remain key challenges. This review aims to provide valuable insights and guidance for the continued advancement of SACs to optimize their practical applications and accelerate their integration into large-scale catalytic processes.
-
Keywords:
- single atom /
- catalyst /
- asymmetric coordination /
- electrocatalysis /
- control strategy
-
随着全球能源消耗的增加和气候变暖,开发高效、低成本、稳定的电催化剂对现代社会的可持续发展至关重要[1−6]. 2011年,Qiao等[7]首先提出了“单原子催化”的概念. 单原子催化剂(SACs)具有独特的电子和几何结构、接近100%的原子利用率、理想的催化活性、优异的产物选择性和稳定性等优点[8−9]. 因此,它在电催化、光催化、酶催化等许多领域得到了迅速的发展[10−13]. 目前,研究人员正致力于调节SACs的各种条件以增强其催化活性和稳定性,如缺陷工程,载体效应,配位微环境调控等[14−17]. 研究表明,适当的金属原子及其局部配位结构的调控对催化反应有决定性作用. 当前SACs配位微环境的调控策略有改变配位物种,调节配位数,杂原子引入以及引入其他金属等策略[18−20].
催化剂的催化效率在很大程度上取决于两个要素,即活性位点的数量和单个活性位点的本征活性. 其中,M—Nx位点是公认的碳基SACs的活性中心. 在1964年,Jasinski[21]成功制备了Co—N4配位的酞菁钴催化剂,并发现它能在碱性条件下进行氧还原反应(ORR). 目前,酞菁、卟啉和其他具有取代官能团的衍生物已被用于制备具有四配位的过渡金属中心(Fe、Co、Ni、Cu等)的大环分子[22−26]. 然而,有报道指出M—N4催化剂因其强电负性直接影响了反应中间体在金属活性位点表面吸附的吉布斯自由能,减慢了催化反应的动力学过程[27−28]. 在催化反应过程中,以二氧化碳还原反应(CO2RR)为例,具有高电负性N原子的对称配位构型可能导致中间体的吸附强度不合适,从而减慢整个反应过程. 当CO2还原为CO时,*COOH是CO转化的关键中间体,而CO*中间体则与CO的解吸有关. 虽然表现出较低的起效电位,但已证明CO*在Fe—N4位点上的强吸附,导致选择性和稳定性不理想[29]. 对于上述问题,研究人员发现通过对SACs配位结构的合理调整,可以设计出具有理想催化性能的SACs. 例如,Li等[30]以锌基沸石咪唑骨架(ZIF‒8)、聚丙烯腈(PAN)、双氰胺为原料,成功地制备了具有轴向氮配位的Fe—N5位点的Fe SACs,其合成的催化剂具有过电位低、高法拉第效率、稳定性好等特点. 密度泛函理论(DFT)证实了优异的CO2RR性能是轴向氮配位的特殊结构,轴向配位增强了电子的局域化,从而降低了CO2RR的能垒,抑制了析氢反应(HER),具有较好的活性和选择性.
近年来,研究人员通过调整金属前驱体、氮源、碳源的种类和比例,或加入其他非金属杂原子,制备了多种不对称配位SAC,如:M—N4—Y、M—Nx—Y及M—M等结构的非对称配位SACs. 与传统的M—N4结构的SACs相比,非对称配位结构的SACs通过因其独特的电子和几何结构而受到广泛的研究. 到目前为止,对于非对称配位SACs的研究主要集中于不同构型催化剂的结构及其在各种催化反应中的应用,对于不同构型的SACs的针对性制备缺少统一的规律总结. 本文系统总结了近年来电催化非对称配位SACs的研究进展. 重点研究了M—N4—Y(Y为轴向杂原子)、M—Nx—Y(Y为杂原子)和M—M结构的制备策略,这有助于预测其后续合成. 此外,我们还总探究了催化反应中结构和性能的关系,包括CO2RR、ORR、硝酸根还原反应(NO3RR)和电解水反应(析氧反应OER和析氢反应HER). 最后,对非对称配位SACs的发展提出了展望. 我们希望本综述能够为非对称配位单原子催化剂的研究提供一些进步的观点,并有助于制备高效稳定的非对称配位单原子催化剂.
1. 单原子催化剂的非对称配位结构的合成策略
到目前为止,具有不对称电子构型的非对称配位SACs,由于其中心金属原子独特的电子填充状态和电荷分布,从而可以改善催化剂与反应物之间的相互作用[31]. 目前,在大多数的研究中,以M—N4—Y(Y为轴向杂原子),M—Nx—Y(Y为杂原子)和M—M配位构型为主[32−35]. 在本章节中,着重介绍这三种配位构型的合成策略.
1.1 M—N4—Y(Y为轴向杂原子)
轴向配位策略最早由天然酶如辣根过氧化物酶(HRP)和细胞色素C氧化酶(CcO)的活性铁位点得到启发. Xu等[36]以石墨烯作为基体,双氰胺为氮源,通过热解法合成出具有五配位血红素样活性位点结构(Fe—N5—C)的Fe基SACs在电催化剂. 轴向配位的第5个单原子位点的推力效应赋予了Fe活性位点与中间体更强的相互作用,增加了活化H2O2的能力. 与传统的M—N4型SACs相比,在SACs的平面M—N4位点上引入垂直、非共面轴向的配体,可以破坏其电子分布的对称性,有效的改变中心金属原子活性位点的电子结构,从而优化中间体吸附和脱附过程,进而改变反应途径[37−39]. 目前,轴向杂原子的配体有O、P、S、卤素等[40−43].
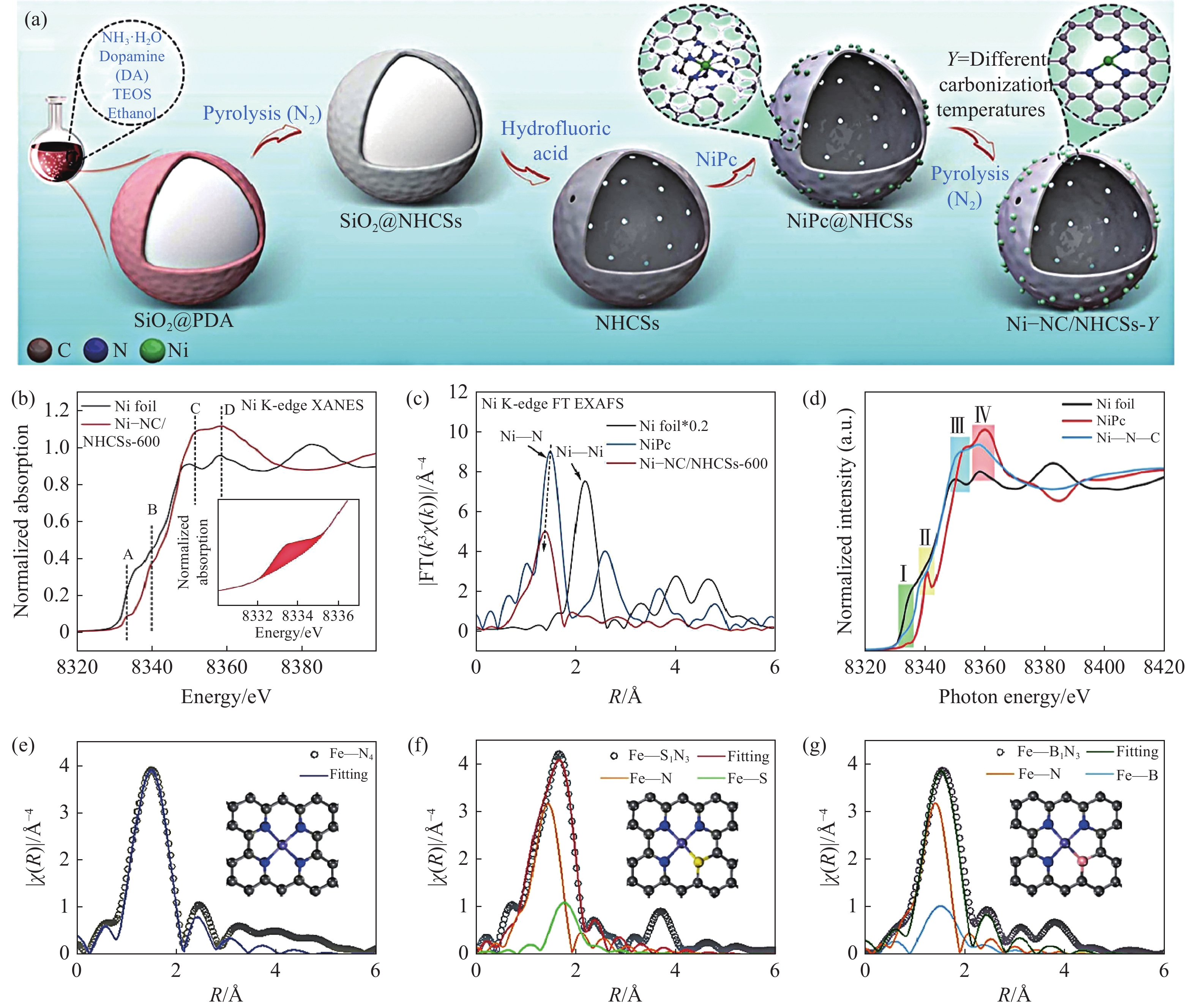
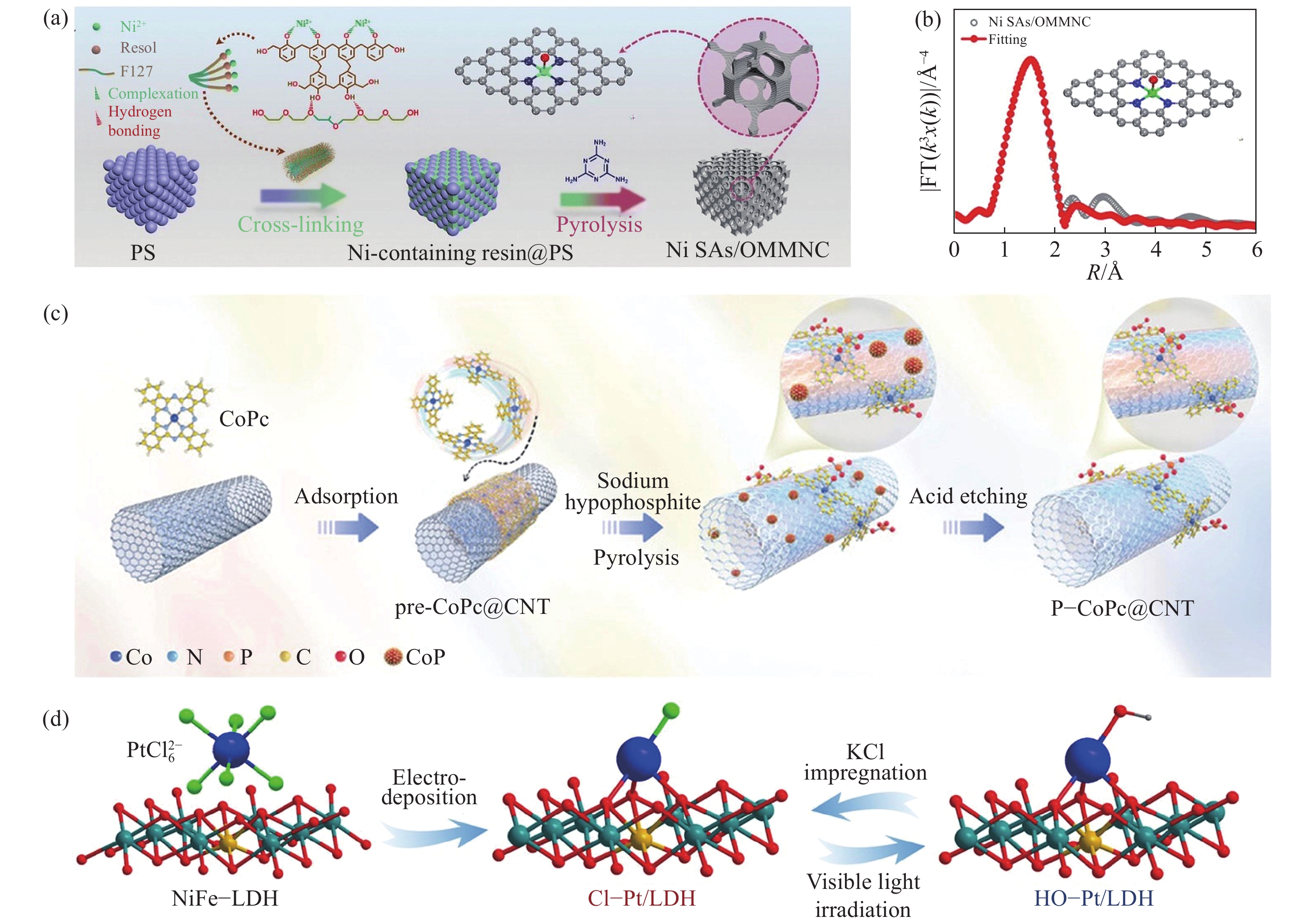
由于O和N的电负性不同,在传统的M—N4结构中引入轴向O原子可以打破M—N4的对称电子结构,从而提高电催化性能. 通常,合成具有轴向氧配位的SACs需要富O前体[44]. 如图1(a)所示,Li等[45]使用可溶性酚醛树脂溶胶作为富O前驱体与Ni(NO3)2在乙醇中溶解形成Ni2+络合溶胶,再与合成的大孔模板热交联,在900 ℃下和三聚氰胺一起热解,随后通过酸刻蚀去除模板得到轴向氧配位的Ni—N4构型的催化剂(Ni SAs/OMMNC). X射线光电子能谱(XPS)分析揭示了Ni SAs/OMMNC的化学成分和化学状态. 其中,Ni SAs/OMMNC的高分辨率N 1s光谱存在Ni—N(399.3 eV),证明了金属与氮配位成键. 另一方面,对Ni SAs/OMMNC中Ni K-edge进行X射线吸收近边结构(XANES)光谱和EXAFS光谱分析进一步证明Ni原子的电子结构和配位环境. XANES光谱显示,表明Ni SAs的价态在0到+2之间. 此外,Ni SAs/OMMNC和NiPc之间相似的特征进一步表明Ni SAs/OMMNC中存在方形平面的Ni—N4基团(图1(b)). Ni SAs/OMMNC的傅里叶变换k3加权EXAFS谱(FT‒EXAFS)在1.49 Å附近有一个优势峰,这归因于Ni—N/Ni—O键的第一个配位壳且没有发现典型的Ni—Ni配位峰. 此外,小波变换光谱呈现出Ni—N/Ni—O路径的单一强度最大值,约为4.7 Å,与NiPc和NiO接近,进一步证实了Ni—N/Ni—O配位的存在. 除此之外,Wang等[46]以氧化石墨烯为富氧基质,通过范德华相互作用合成O—Co—N4构型的催化剂. 另一方面,高温热解法作为常见的催化剂合成方法,也可制备出轴向氧配位的SACs. Wang等[47]通过直接碳化前驱体,然后酸蚀,合成了O轴配位的Ni—N4—O/C催化剂. 具体来说,以Mn基金属有机骨架为活性位点主体合成Ni—N4—O/C前驱体(Mn, Ni‒MOFs),然后原位引入醋酸镍作为客体,在氨气氛和酸浸处理下进一步碳化后,O原子被锚定在碳基体内的Ni—N4部分上. 同样的,与O同一主族元素S也被广泛用于轴向杂原子. Hu等[48]采用巯基咪唑和二甲基咪唑的混合配体合成了含有巯基的ZIF‒8(SH‒ZIF‒8),将SH‒ZIF‒8进入植酸掺杂P元素后在
1000 ℃下煅烧得到ZnN4S1/P‒HC. 通过高分辨率S 2p XPS光谱证明了Zn—S键合的存在. 同时,P 2p谱揭示了P原子被掺杂到碳骨架中,而不与Zn原子配位. 同时,同步X射线吸收光谱测量结果显示经过傅里叶变换扩展Zn K-edge的EXAFS显示了一个主峰,与酞菁锌(ZnPc)中的Zn—N峰相近,且未检测到Zn—Zn和Zn—O的配位,这排除了Zn或ZnO聚集的形成. 进一步通过R和q空间的EXAFS曲线拟合分析,研究了Zn在ZnN4S1/P‒HC中的配位构型. 结合XPS和XANES结果从而确定了一个ZnN4S1模型,其中S配位在轴向位置,与相邻的S掺杂石墨烯层形成配位. DFT计算结果验证了轴向S的形式,并确定了ZnN4S1/P‒HC是最佳的模型.![]() 图 1 轴向配位的SACs合成. (a) Ni—N4—O/C的合成工艺说明[45]; (b) Ni—N4—O/C在R空间中的EXAFS拟合曲线,插图为Ni—N4—O/C相应的示意图模型[45]; (c) P‒CoPc@CNT合成示意图[39]; (d) 电沉积法合成Cl—Pt/LDH催化剂及可逆轴向配体交换过程示意图(原子由球体表示:Pt (蓝色),Ni (橄榄色),Fe (黄色),O (红色),Cl (绿色)和H (灰色))[49]Figure 1. Synthesis of SACs with axial coordination: (a) schematic illustration of the synthesis process of Ni—N4—O /C[45]; (b) Ni—N4—O/C EXAFS fitting curve in R-space, with the inset showing the corresponding schematic model of Ni—N4—O /C[45]; (c) P‒CoPc@CNT composite diagram[39]; (d) schematic of synthesis of Cl—Pt/LDH catalyst by electrodeposition and reversible axial ligand exchange process (Atoms are represented by spheres: Pt (blue), Ni (olive), Fe (yellow), O (red), Cl (green), and H (gray)) [49]
图 1 轴向配位的SACs合成. (a) Ni—N4—O/C的合成工艺说明[45]; (b) Ni—N4—O/C在R空间中的EXAFS拟合曲线,插图为Ni—N4—O/C相应的示意图模型[45]; (c) P‒CoPc@CNT合成示意图[39]; (d) 电沉积法合成Cl—Pt/LDH催化剂及可逆轴向配体交换过程示意图(原子由球体表示:Pt (蓝色),Ni (橄榄色),Fe (黄色),O (红色),Cl (绿色)和H (灰色))[49]Figure 1. Synthesis of SACs with axial coordination: (a) schematic illustration of the synthesis process of Ni—N4—O /C[45]; (b) Ni—N4—O/C EXAFS fitting curve in R-space, with the inset showing the corresponding schematic model of Ni—N4—O /C[45]; (c) P‒CoPc@CNT composite diagram[39]; (d) schematic of synthesis of Cl—Pt/LDH catalyst by electrodeposition and reversible axial ligand exchange process (Atoms are represented by spheres: Pt (blue), Ni (olive), Fe (yellow), O (red), Cl (green), and H (gray)) [49]P,另一种轴向杂原子配位元素,已经被用于SACs的结构修饰且表现出较好的催化活性. Liu等[39]将酞菁钴通过π‒π相互作用预吸附在碳纳米管(预CoPc@CNT)上,然后与过量的次磷酸钠混合之后,将混合物在300 ℃下热解2 h,然后通过酸蚀去除磷化物(图1(c)). 在此温度下,次磷酸钠分解成${\mathrm{PO}}_4^{3-} $,而CoPc和CNT能够保留原始结构,最终形成PO4为轴向配位的Co—N4构型的高OER性能催化剂(P‒CoPc@CNT). 在P 2p的XPS光谱中,P‒CoPc@CNT在134.8 eV处显示单个P 2p峰,这归因于${\mathrm{PO}}_4^{3-} $中深度氧化的P原子,O 1s XPS显示P—O(533 eV)和P—O—Co(桥O原子)(531.5 eV)共存,表明O的化学环境来源于PO4轴向配位. 此外,用Co K-edge XAFS实验验证了PO4与Co1N4单原子的轴向配位. 在EXAFS光谱中,P‒CoPc@CNT两个特征峰对应于1.92和2.49 Å,与平面Co—N和轴向Co—O高度一致. 基于EXAFS的拟合提供了P‒CoPc@CNT具体配位数(CN). 平面Co—N键的CN为3.95,结合轴向Co—O键的CN为1.24,验证了O2P—O—Co1N4或[O3P—O—CoPc]2−在轴向PO4中的局部结构.
除去O、S和P元素作为轴向配位元素外,卤素为轴向配位的引入更为常见. Zhang等[49]以NiFe‒LDH纳米片阵列为载体,采用电沉积法合成了Cl‒Pt/LDH催化剂. 如图1(d)所示,以NiFe‒LDH为工作电极,在含${\mathrm{{{PtCl}}}}_6^{2-} $阴离子的1 mol·L−1 KOH溶液中,采用三电极体系制备了Cl‒Pt/LDH. 在电解过程中,${\mathrm{{PtCl}}}_6^{2-} $阴离子被电沉积并吸附在LDH表面,生成具有轴向Cl配位的原子分散Pt位. 而轴向配体可以通过进一步的光化学方法改变. 在可见光照射下,轴向的Cl配体被氢氧根取代. 此外,经过KCl浸渍处理后,交换的羟基可以可逆地切换回轴向Cl配体.
总的来说,当前轴向配位SACs的合成方法有很多,高温热解是最常用的方法. 然而,由于高温下反应过程的复杂机制,对于准确的合成目标催化剂的还存在一定的挑战. 因此,需要理解合成过程中机制及对可控合成的方法进行探究,以制备出具有普适性的轴向配位的SACs.
1.2 M—Nx—Y(Y为杂原子)
制备非对称配位结构SACs的其中一种策略是通过调节合成条件,在同一平面内形成中心金属原子与三个氮原子和一个杂原子配位结构. 据研究报道结果表明,通过合理调节热解温度,可以得到不同配位数的SACs[50−51]. Gong等[52]通过调节热解温度,制备了一系列不同N原子配位数的Ni—NC位点用于CO2RR. 其中,热解温度分别为600、800和900 ℃,得到的催化剂配位分别为Ni—N4—C、Ni—N3—C和Ni—N2—C,温度越高Ni SACs中的N的配位数越少. 此外,高温热解过程中存在的热应力、化学反应及气体刻蚀等原因,可以制备出具有低配位缺陷的不对称配位结构的催化剂[53−54]. 去除部分配位的氮原子后,形成的低配位M—Nx结构的催化剂不仅会破坏传统对称的M—N4结构,还会出现缺陷[55−56]. 如图2(a)所示,Gong等[57]通过自组装配位技术和高温热解策略合成出具有缺陷的Ni—N3结构催化剂(Ni–NC/NHCSs-600). 首先,研究人员通过模板法合成具有聚多巴胺层的SiO2球,在镍盐中搅拌,干燥后分别在800 ℃下热解形成Ni—N3配位结构. 由于Ni—O键的强度低于Ni—N键,Ni—O随着热解温度的升高导致不对称的Ni—N3和相邻的氮位空缺. 通过XPS光谱显示,热解后的Ni–NC/NHCSs-600中N、Ni元素的含量均降低了,这说明热处理后Ni原子和—CN片段丢失. —CN片段的丢失会导致金属种化学态的波动,—CN片段被剪切,导致一定比例的Ni—N键丢失,可能在Ni原子周围形成氮空位结构[58],从而使孤立的Ni原子的配位数降低,导致Ni原子的电子云增加[58−59],使价态向结合能较低的方向移动,同时使Ni的氧化态降低. Ni原子的XANES和EXAFS结果进一步证明了Ni—N3配位结构和缺陷的存在(图2(b)和图2(c)). 在Ni K-edge XANES光谱中发现明显的前边缘峰,表明偶极允许跃迁增加(1s→4p),其中Ni原子的4p轨道与3d轨道杂化[15]. Cabaret等[60]证明了这种偶极子转变只发生在非中心对称结构上,说明Ni–NC/NHCSs-600中Ni原子的配位环境不同于传统对称结构的Ni—N4. 在Ni–NC/NHCSs‒600中发现了约
8340 eV的弱峰B(1s到4pz跃迁),进一步说明Ni–NC/NHCSs-600中Ni原子的D4h对称变形可能是由Ni–NC/NHCSs-600中不饱和的Ni配位结构引起的. EXAFS结果表明与传统的Ni—N4对称结构不同的是Ni物种的配位数为2.9±0.4,表明中心Ni原子周围有三个N原子,形成了不饱和的配位结构,且中心Ni原子与N原子之间的键长比NiPc中的Ni—N键长更短,进而证明了Ni—N3配位构型和缺陷的存在. 同样的,Zhou等[61]以葡萄糖为碳源,双氰胺为氮源,与镍盐混合、干燥,通过两步热解法分别在900 ℃和700 ℃下煅烧得到具有Ni—N2配位和氮缺陷的协同的催化剂(Ni—N—C). Ni—N—C和NiPc的不同特征(Ⅰ‒Ⅳ)表明它们具有不同的局部协调环境(图2(d)). Ni—N—C的3d‒4p轨道杂化导致的特征I不允许电偶极子,但允许四极子中的1s‒3d跃迁,这种特征在Ni—N—C中强于NiPc. 这一结果表明Ni—N—C中Ni位的局部配位结构是高度无序/缺陷的. 此外,Ni—N—C的特征Ⅱ(对应于D4h构型中偶极允许的1s−4pz跃迁)明显弱于NiPc,这也表明Ni—N—C中Ni位点的结构不对称. 特征Ⅲ与特征Ⅳ的强度比(分别对应于1s−4px、y跃迁和多次散射过程)代表了Ni位点的显著位移,这进一步反映了结构不对称的存在.![]() 图 2 杂原子掺杂的SACs合成. (a) Ni—NC/NHCSs-Y合成示意图[57]; (b) Ni–NC/NHCSs-600和Ni箔的Ni K-edge XANES光谱,插图为Ni–NC/NHCSs-600的扩展前边缘区域(峰A)[57]; (c) k3加权傅立叶变换EXAFS光谱(FTEXAFS)[57]; (d) Ni—N—C、Ni箔和NiPc的Ni K-edge XANES光谱[61]; (e‒g) Fe—N4,FeS1N3和FeB1N3在R空间中的EXAFS拟合曲线,插图为相应的示意图模型[63]Figure 2. Synthesis of SACs doped with heteroatoms: (a) schematic illustration of Ni—NC/NHCSs-Y synthesis[57]; (b) Ni K-edge XANES spectra of Ni–NC/NHCSs-600 and Ni foils, with the inset showing extended front edge region of Ni–NC/NHCSs-600 (Peak A)[57]; (c) k3 weighted Fourier transform EXAFS spectrum (FTEXAFS)[57]; (d) Ni K-Edge XANES spectra of Ni—N—C, Ni foil, and NiPc[61]; (e‒g) EXAFS fitted curves of Fe—N4, FeS1N3, and FeB1N3 in R-space, respectively. The insets depict the corresponding schematic models[63]
图 2 杂原子掺杂的SACs合成. (a) Ni—NC/NHCSs-Y合成示意图[57]; (b) Ni–NC/NHCSs-600和Ni箔的Ni K-edge XANES光谱,插图为Ni–NC/NHCSs-600的扩展前边缘区域(峰A)[57]; (c) k3加权傅立叶变换EXAFS光谱(FTEXAFS)[57]; (d) Ni—N—C、Ni箔和NiPc的Ni K-edge XANES光谱[61]; (e‒g) Fe—N4,FeS1N3和FeB1N3在R空间中的EXAFS拟合曲线,插图为相应的示意图模型[63]Figure 2. Synthesis of SACs doped with heteroatoms: (a) schematic illustration of Ni—NC/NHCSs-Y synthesis[57]; (b) Ni K-edge XANES spectra of Ni–NC/NHCSs-600 and Ni foils, with the inset showing extended front edge region of Ni–NC/NHCSs-600 (Peak A)[57]; (c) k3 weighted Fourier transform EXAFS spectrum (FTEXAFS)[57]; (d) Ni K-Edge XANES spectra of Ni—N—C, Ni foil, and NiPc[61]; (e‒g) EXAFS fitted curves of Fe—N4, FeS1N3, and FeB1N3 in R-space, respectively. The insets depict the corresponding schematic models[63]在形成的非对称配位结构的SACs催化剂中,也可用一个杂原子(P,B,S等)取代一个N原子进而形成不对称配位的M—N3—Y(Y为杂原子)催化剂[62]. Jin等[63]提出在合成前驱体中分别加入硫粉和硼酸,通过900 ℃的高温热解分别合成了Fe—S1N3和Fe—B1N3配位构型的Fe SACs催化剂. EXAFS光谱结果证实FePc和Fe—N4中Fe K-edge FT‒EXAFS光谱在约1.47 Å处呈现主峰,对应于Fe—N路径(图2(e)). 在Fe—S—N3和Fe—B—N3中,该峰分别位移到约1.66 Å和约1.55 Å,表明由于S和B的引入,第一壳层配位环境发生了变化(图2(f)和图2(g)). 除此之外,S原子取代其他原子的策略也被用于构建镍基、锰基的不对称配位SACs,这些SACs在CO2RR和电化学水分解中具有增强的反应活性. Yang和他的同事[64]通过煅烧${\mathrm{SO}}_4^{2-} $掺杂Zn/Ni ZIF,合成了一种新型的S/N共配Ni单原子电催化剂(Ni‒SNC),用于电催化CO2RR. 利用像差校正的HAADF‒STEM来确定了Ni‒SNC催化剂中存在单原子Ni. S与归一化后的XANES光谱证明了Ni‒SNC中Ni的平均价态在+1至+2之间. EXAFS光谱的傅里叶变换显示,Ni‒SNC催化剂没有明显的Ni—Ni峰. Ni—N平均配位数为2.9,Ni—S的配位数为1.3,从而确定了催化剂结构为Ni—N3S.
研究表明,在M—Nx结构中加入P原子可以通过增强C原子周围的电荷离域效应来改善SACs的性能. 其中,三苯基膦(PPh3)作为磷源被广泛应用于含磷催化剂的合成. Xu等[65]通过原位磷化‒吸附‒热解策略合成了具有NO3RR性能的Fe—N3P配位结构的催化剂(Fe—N/P—C). 具体来说,研究人员首先在传统合成ZIF‒8结构的方法过程中加入PPh3作为P源以合成ZIF‒8‒P,并将ZIF‒8‒P在含有Fe盐的溶液中超声、搅拌合成Fe/ZIF‒8‒P,在950 ℃下热解以除去Zn元素最终制备出具有Fe—N3P配位结构的SACs. 根据N的XPS光谱显示,在398.8 eV处存在Fe—N峰. 而在P的2p XPS光谱中,除了P—C(132.4 eV)和P—N(133.3 eV)两种峰,还有P 2p3/2(128.4 eV)和P 2p1/2(129.1 eV)特征峰,这是由Fe—P键的形成衍生而来用P取代部分N原子的位置. 同样的,EXAFS光谱显示,Fe—N/P—C和Fe—N—C分别在1.50 Å和1.53 Å处有一个明显的傅立叶变换峰这可以归结为Fe和光原子的后向散射. 结果表明,与Fe—N—C相比Fe—N/P—C的主峰强度较弱,这是由于P部分取代了N,打破了第一配位壳层中的原始Fe—N4局部结构. Wan等[66]使用相同方法合成了Co—N3P配位结构的催化剂. 首先,研究人员采用Zn/Co双金属MOF作为模板和前体,利用其内部的分子尺度笼包封磷源(PPh3). 通常情况下,PPh3与双金属MOF的前体(Zn2+,Co2+和2-甲基咪唑)混合. 自组装过程后,PPh3分子被捕获在MOF笼中(表示为P@Zn/Co‒MOF). 然后将P@Zn/Co‒MOF在950 ℃氩气条件下热解得到Co‒SA/P.
除了N、S和P之外,碳载体上O的配位作用能够有效调节金属原子的d带中心位置,并影响金属d带相对于费米能级的能量分布. 这种调节使得反应中间体在金属表面的吸附和解吸过程变得更加容易,从而有利于催化反应的进行[67]. Li等[68]通过水热法和热解法合成Fe—N3O配位的SACs. 在合成过程中,甲酰胺提供氮源,将Fe盐、Zn盐和乙二醇混合,在180 ℃下进行水热反应,得到具有金属和N,O配位结构(Fe/Zn)的单原子前驱体. 先前的研究报道中,甲酰胺小分子可以通过席夫碱反应进行缩合并用于螯合金属离子来制备M—N—C材料. 将上述反应用于制备单原子Fe—N—C催化剂. 最重要的是,研究人员额外加入乙二醇分子并参与甲酰胺自缩合,最终在950 ℃下制备了Fe—N3O构型的催化剂.
1.3 M—M
追求高性能催化剂的常见策略是减小负载型多相催化剂中活性金属的尺寸,以最大限度地提高利用效率. 前沿研究发现了原子分散的催化剂(例如SACs、双原子催化剂(DACs)),它们具有许多有趣的催化特性,超越了限制传统催化剂的简单缩放定律[69−71]. DACs的发现从甲烷单加氧酶(70年代)和经典的Cu‒ZSM‒5催化剂(80年代)发展而来,甚至早于单原子催化的发现. 目前,双原子催化剂类型可分为同核双金属和异核双金属[72−73].
1.3.1 M1—M1同核双金属SACs
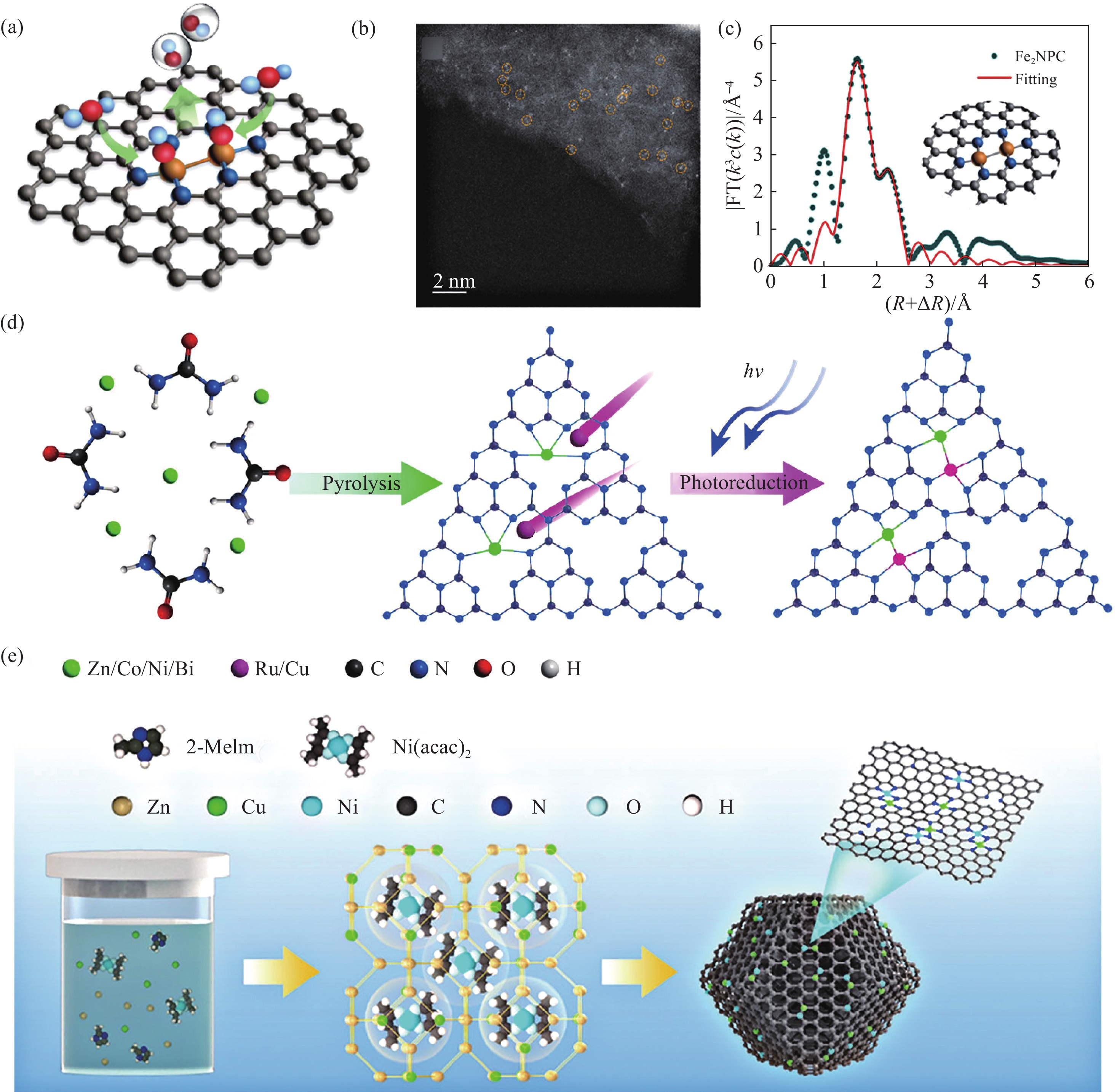
双金属位SACs的研究有助于深入探索其原子尺度结构与催化活性的关系[74−76]. 现有的研究已经证实,设计良好的双原子团簇可以用作金属前体,以构建均匀和精确的双核金属催化剂[77]. Zhao等[78]通过一步热解合成了Fe2—N6构型的DACs,如图3(a)所示. 在ZIF‒8的合成过程中加入了具有本征Fe—Fe键的Fe2(CO)9. 得益于Fe2(CO)9(直径约9.5 Å)和ZIF‒8空腔(空腔大小为11.6 Å,孔隙尺寸为3.4 Å)的尺寸匹配策略,铁前驱体可以被完美地封装在ZIF‒8的空腔中,在氮气气氛中
1000 ℃热解得到Fe2NPC催化剂. HAADF‒STEM图像中显示Fe原子是以原子形式分散的,且是成对存在的(图3(b)). FT‒EXAFS谱图显示在1.62 Å存在Fe—N键,在2.19 Å处存在Fe—Fe键,证实了双原子Fe位的存在(图3(c)). 目前,同核DACs的合成通常采用复杂的双核金属配合物,需要探究更方便、有限的同核DACs合成方法[79−81].![]() 图 3 双金属位点的SACs合成. (a) Fe2NPC结构示意图[78]; (b) Fe2NPC的HAADF‒STEM图像[78]; (c) Fe2NPC的EXAFS R空间拟合曲线[78]; (d) 制备ZnRu‒、NiRu‒、ZnCu‒、CoCu‒、NiCu‒和BiCu‒PCN异核DACs的导航定位策略示意图[82]; (e) Ni/Cu—N—C制备原理[86]Figure 3. Synthesis of bimetallic atomic sites: (a) Fe2NPC structure diagram[78]; (b) HAADF‒STEM images of Fe2NPC[78]; (c) Fe2NPC EXAFS R-space fitting curve[78]; (d) preparation of ZnRu‒, NiRu‒, ZnCu‒, CoCu‒, NiCu‒, and BiCu‒PCN heteronuclear DACs navigation strategy diagram[82]; (e) Ni/Cu—N—C preparation principle[86]
图 3 双金属位点的SACs合成. (a) Fe2NPC结构示意图[78]; (b) Fe2NPC的HAADF‒STEM图像[78]; (c) Fe2NPC的EXAFS R空间拟合曲线[78]; (d) 制备ZnRu‒、NiRu‒、ZnCu‒、CoCu‒、NiCu‒和BiCu‒PCN异核DACs的导航定位策略示意图[82]; (e) Ni/Cu—N—C制备原理[86]Figure 3. Synthesis of bimetallic atomic sites: (a) Fe2NPC structure diagram[78]; (b) HAADF‒STEM images of Fe2NPC[78]; (c) Fe2NPC EXAFS R-space fitting curve[78]; (d) preparation of ZnRu‒, NiRu‒, ZnCu‒, CoCu‒, NiCu‒, and BiCu‒PCN heteronuclear DACs navigation strategy diagram[82]; (e) Ni/Cu—N—C preparation principle[86]1.3.2 M1—M2异核双金属SACs.
M1—N4和M2—N4活性位点掺杂及M1—M2—Nx催化剂与M1—M2金属键是异核双金属位催化剂的主要形式,在电催化领域得到了广泛的研究. Zhao等[82]提出了一种“导航和定位”策略,在聚合物氮化碳(PCN)上精确和可扩展地合成一系列异核M1—M2异核双金属SACs(ZnRu‒,NiRu‒,ZnCu‒,CoCu‒,NiCu‒和BiCu‒PCN). 如图3(d)所示,以ZnRu‒PCN为例,首先通过在空气中500 ℃煅烧尿素和Zn(OOCCH3)2来合成Zn‒PCN SAC,以形成Zn成核位点. 在光激发下,Zn‒PCN中的Zn位可以接受来自光敏化PCN载体的光致电子产生电荷富集中心,从而吸引第二Ru3+离子到Zn位. 最终Ru离子被还原并锚定在Zn位点周围,通过RuCl3的原位光还原形成异核ZnRu‒PCN DAC. 其中,光激发是构建异核ZnRu‒PCN DAC的必要条件,在Zn中心周围形成Zn—Ru二聚体的电子富集. HAADF‒STEM图像显示,在ZnRu‒PCN DAC中PCN载体上明显检测到许多双原子位点,其M1—M2距离约为2.5 Å. 此外,原子分辨电子能量损失谱(EELS)也清楚的证实了ZnRu‒PCN中存在一个Zn和一个Ru原子,有力地证明了M1M2‒PCN DACs中异核双原子对的形成. 同样的,ZnRu‒PCN DAC的FT‒EXAFS拟合结果揭示了ZnRu‒PCN中Ru和Zn中心的RuN3和ZnN3结构.
除上述光激发诱导合成策略外,制备DACs最常用的方法为高温热解金属盐的各种载体或金属有机前驱体. 例如,Fe掺杂ZnCo‒ZIF,FeCo‒酞菁‒ZIF‒8和多巴胺包覆钴镍金属有机骨架分别被热解合成FeCo和CoNi DAC[83−85]. 此外,将Fe3+掺入Ni掺杂的聚多巴胺中,将Zn2+和Co2+的混合金属盐引入壳聚糖中,通过热解和酸处理构建DACs[16−17]. 这种方法合成的DACs,其金属配位通常是相同的. 例如图3(e)所示,Cheng等[86]在合成ZIF‒8过程中加入镍盐和铜盐已形成Ni/Cu‒ZIF,在
1100 ℃下热解直接得到Ni/Cu—N—C催化剂. 在FT‒EXAFS图谱中,Ni/Cu—N—C中的Ni和Cu元素分别存在Ni—N(1.4 Å)、Cu—N(1.5 Å)路径. EXAFS拟合结果证明了Ni和Cu原子的配位数分别为4.4和4.3,即NiN4和CuN4配位构型. Ni/Cu—N—C异核双金属的协同作用,促进了*COOH中间体的吸附,从而促进了CO的生成.2. 非对称配位单原子催化剂的应用研究
SACs因其成本效益高、原子利用效率最大化和结构可调等优点,在电催化中的应用已成为一个重要研究内容. 金属活性位点的固有活性和密度是影响SACs催化能力的关键因素. 因此,人们设计和研究了具有不同活性位点和不同配位结构的SACs. 通过大量研究发现,精确调控金属原子周围的微环境结构可以显著提高催化剂的电化学性能. 到目前为止,为了进一步提高SACs的电化学性能,研究人员提出并实施了几种策略,如缺陷策略,杂原子策略,轴向配位策略等. 通过引入或减少中心金属原子的配位数,从而改变中心原子的配位环境和电子结构,进而调节SACs的电催化性能. 在本章节中,我们将重点讨论上一章节中非对称配位结构的SACs构型在各种电化学反应中的电催化性能规律总结.
2.1 M—N4—Y(Y为轴向杂原子)
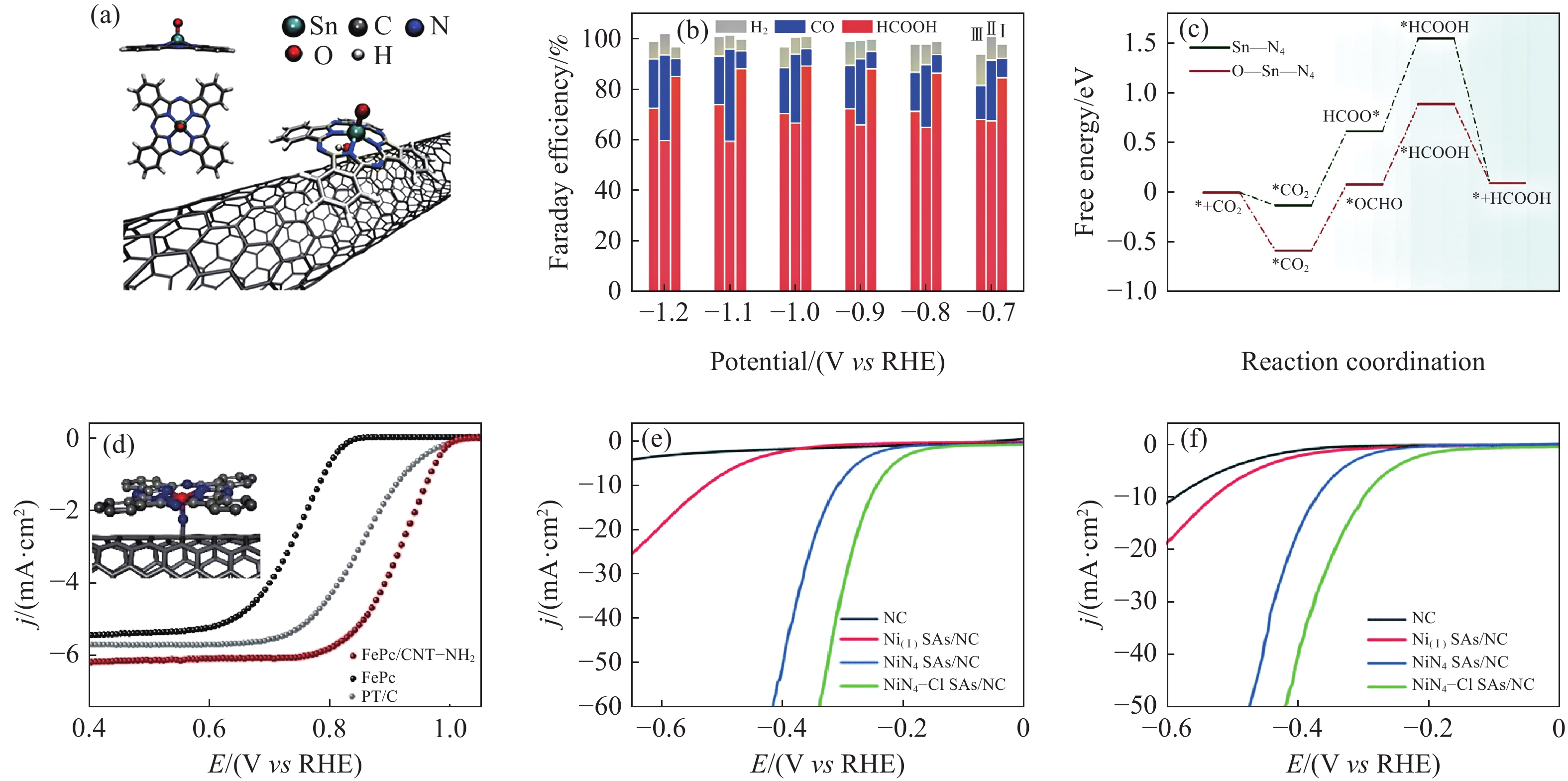
在将CO2通过电化学CO2RR产生CO过程中,在高CO法拉第效率(FECO)下的狭窄电势范围仍然阻碍了其最终的实际可行性. 通过杂原子掺杂来修饰电子结构被认为是有效的,这一方法有效的原因主要归因于以下几点,(1)调节电子密度:杂原子的引入可以改变SACs中金属原子周围的电子密度. 这种电子密度的变化可以影响金属原子与反应中间体之间的相互作用,从而优化催化性能. (2)新配位键的形成:杂原子可以与中心金属原子形成新的配位键,这些配位键可以改变金属原子的电子结构和化学性质,进而影响催化剂的催化活性. Chen等[87]通过快速热解和可控活化策略,构建了富O碳质载体和具有轴向O配位的原子分散的FeN4位点(Fe1N4—O1),并通过像差校正电子显微镜和X射线吸收光谱证实了这一点. 当FECO连续保持在接近100%时,可以实现310 mV(‒0.56 V至‒0.87 V vs RHE)的宽电位范围. DFT计算表明,轴向氧的配位增强了Fe活性位点的电子局域化,导致Fe‒3d轨道的d带中心向较低能量水平移动,从而适度的调整了CO2RR中间体结合的相互作用,进而提高了CO2RR的选择性以及实现宽电位范围内的高CO的法拉第效率. 同样的,Wang等[47]通过对锰金属有机骨架的热解、酸浸合成出具有轴向氧配位构型的Ni—N4催化剂(Ni—N4—O/C). Ni—N4—O/C催化剂表现出优异的电化学CO2RR性能,在‒0.9 V时,CO的法拉第效率接近100%,在‒0.5 V~‒1.1 V的宽电位范围内,CO的法拉第效率可以保持在90%以上. 这一优异性能的表现归因于轴向氧原子的引入导致了电子的推移效应,增强了Ni—N4—O位点的电荷极化,从而优化了表面状态,降低了CO2分子到*COOH转化过程中具有较低的势垒. 当前研究结果表明,Sn元素在CO2RR中通常更倾向于甲酸的产生. 如图4(a),Deng等[88]设计出具有轴向氧结构的Sn—N4催化剂(O—Sn—N4). 在‒1.0 V vs RHE下,当偏电流密度(jHCOOH)为74.8 mA·cm−2时,CO2在Sn单原子位点上还原为甲酸的选择性和活性与氧轴向配位的Sn(IV)—N4基团(O—Sn—N4)相关,达到了89.4%的最佳HCOOH法拉第效率(图4(b)). 在CO2RR过程中,O—Sn—N4上的第一个CO2‒to‒*CO2台阶的下坡能垒为‒0.58 eV,而Sn—N4上的下坡能垒为‒0.13 eV,表明CO2在O—Sn—N4上更容易发生活化(图4(c)). 此外,O—Sn—N4在形成*HCOOH时表现出相对较低的能垒(ΔG = 0.8 eV),比Sn—N4(ΔG = 0.93 eV)更低,这导致O—Sn—N4对CO2RR对HCOOH的电催化活性更高,因而HCOOH具有高法拉第效率. 总的来说,轴向配位的引入实现了催化剂在宽电势范围内的高FECO,同时对不同中心金属种类引入轴向配体可以加快反应中间体的生成.
![]() 图 4 (a) SnPc/CNT−OH的制备示意图[88]; (b) Ⅰ:SnPc/CNT−OH、Ⅱ:SnPc/CNT−NH2和Ⅲ:SnPc/CNT在不同阴极电位下的法拉第效率[88]; (c) O—Sn—N4和Sn—N4位点上CO2RR到HCOOH的吉布斯自由能图[88]; (d) 0.1 M KOH下FePc、FePc/CNT‒NH2和Pt/C的LSV曲线[14]; (e和f) HER极化曲线[92]Figure 4. (a) Schematic illustration of the preparation of SnPc/CNT−OH[88]; (b) Faraday efficiencies of Ⅰ: SnPc/CNT−OH, Ⅱ: SnPc/CNT−NH2, and Ⅲ: SnPc/CNT at different cathode potentials[88]; (c) Gibbs free energy graphs of CO2RR to HCOOH at O—Sn—N4 and Sn—N4 sites[88]; (d) LSV curves of FePc, FePc/CNT‒NH2, and Pt/C at 0.1 M KOH[14]; (e and f) HER polarization curve[92]
图 4 (a) SnPc/CNT−OH的制备示意图[88]; (b) Ⅰ:SnPc/CNT−OH、Ⅱ:SnPc/CNT−NH2和Ⅲ:SnPc/CNT在不同阴极电位下的法拉第效率[88]; (c) O—Sn—N4和Sn—N4位点上CO2RR到HCOOH的吉布斯自由能图[88]; (d) 0.1 M KOH下FePc、FePc/CNT‒NH2和Pt/C的LSV曲线[14]; (e和f) HER极化曲线[92]Figure 4. (a) Schematic illustration of the preparation of SnPc/CNT−OH[88]; (b) Faraday efficiencies of Ⅰ: SnPc/CNT−OH, Ⅱ: SnPc/CNT−NH2, and Ⅲ: SnPc/CNT at different cathode potentials[88]; (c) Gibbs free energy graphs of CO2RR to HCOOH at O—Sn—N4 and Sn—N4 sites[88]; (d) LSV curves of FePc, FePc/CNT‒NH2, and Pt/C at 0.1 M KOH[14]; (e and f) HER polarization curve[92]同样,轴向配位调控在ORR中的表现方面具有很大的前景. 迄今为止,各种轴向配体(N、O、S、卤素等配体)修饰的SACs对ORR性质的调节能力已被研究. 与传统的M—N4结构的催化剂相比较,具有轴向配位的催化剂整体上都展现出更高的ORR活性,其中绝大多数催化剂的ORR理论过电压小于Pt的理论差值,表现出较好的ORR催化性能. 同时研究发现轴向配体的活性调节作用主要是源于配体与中心金属位点形成化学键,重构中心金属位点的电子结构,降低中心金属位点与ORR中间体轨道杂化,促进中间体*OH的脱附,从而提高ORR活性. 此外,研究发现催化剂的活性随着配位Y的电负性降低而增大. Zhang等[14]基于单核酞菁铁负载在含有供电子官能团(FePc/CNT‒R,“R”为—NH2、—OH). 这些官能团充当与Fe位点配位的轴向配体,通过XPS和基于同步辐射的X射线吸收精细结构证实. 如图4(d)所示,实验结果表明,FePc/CNT‒NH2具有最多的电子供体—NH2轴向配体,表现出最高的ORR活性和正起始电位(Eonset=1.0 V vs RHE)和半波电位(E1/2=0.92 V). 这归因于轴向配位的引入增强了Fe位点的电子密度,从而促进了氧的吸附. 除此之外,与N具有不同电负性的杂原子,如O、S、P和Cl,可以影响中心金属原子的电子结构并调节氧结合能[89]. Zhu等[90]通过预氧化策略来建立Fe—O键,用于合成Fe—N4O1单原子结构. 制备的SAFeN4O1‒C催化剂表现出优异的ORR性能,其半波电位比商用Pt/C高30 mV,在0.8 V vs RHE时的动态电流密度是Pt/C的7.4倍. 根据DFT分析结果显示,轴向的氧配位通过调节电子结构,使Fe的d带中心发生负移,从而优化了氧中间体的吸附能,降低了ORR的能垒. 另一方面,Zhang等[91]提出一种静电自主装策略,通过引入具有轴向O配体在MXene纳米片(CoN4‒O/MX)上形成CoN4O1构型来设计五配位Co位点. CoN4‒O/MX具有优异的双功能电催化性能(OER和ORR),总的过电位为0.72 V. 更重要的是,基于CoN4‒O/MX的锌‒空气电池(ZAB)提供了出色的峰值功率密度,具有250 h(500次循环)的超长循环寿命,在柔性器件中具有强大的实用价值. 这归因于O—CoN4的轴向配位可以引起电子离域来调节Co的三维轨道能级. 轨道重排引起的dz2能级的升高和不饱和占位增加了Co中心的路易斯碱度,容易将电子注入ORR中间体,加速还原步骤.
电解水被普遍认为是将可再生能源转化为高纯H2和O2的最重要且最有前途的途径之一. 目前大多数SACs是基于碳质基底的固有缺陷或杂原子配位来锚定孤立的金属原子. 值得注意的是,轴向配位策略能够调节活性位点与反应中间体之间的结合强度,从而增强催化反应的选择性. 在电解水过程中,这有助于提高目标产物的纯度和产率. Li等[92]通过掺杂‒吸附‒热解策略来构建具有轴向Cl配位的NiN4—Cl活性位点催化剂(NiN4‒Cl SAs/NC). 与无轴向配位的NiN4 SAs/NC相比,NiN4‒Cl SAs/NC在碱性和酸性电解质中具有更优异的HER性能,在10 mA·cm‒2下的过电势分别具有243 mV和274 mV(图4(e)和图4(f)). 密度泛函理论计算表明,NiN4‒Cl SAs/NC优异的HER性能源于轴向Cl配位诱导的电子定位增强,这有利于H*中间体的吸附和活化,从而加速了HER过程. 当前,轴向配位SACs在电解水方面的研究很少,未来该调控策略还有很大的研究潜力.
2.2 M—Nx—Yx(Y为杂原子)
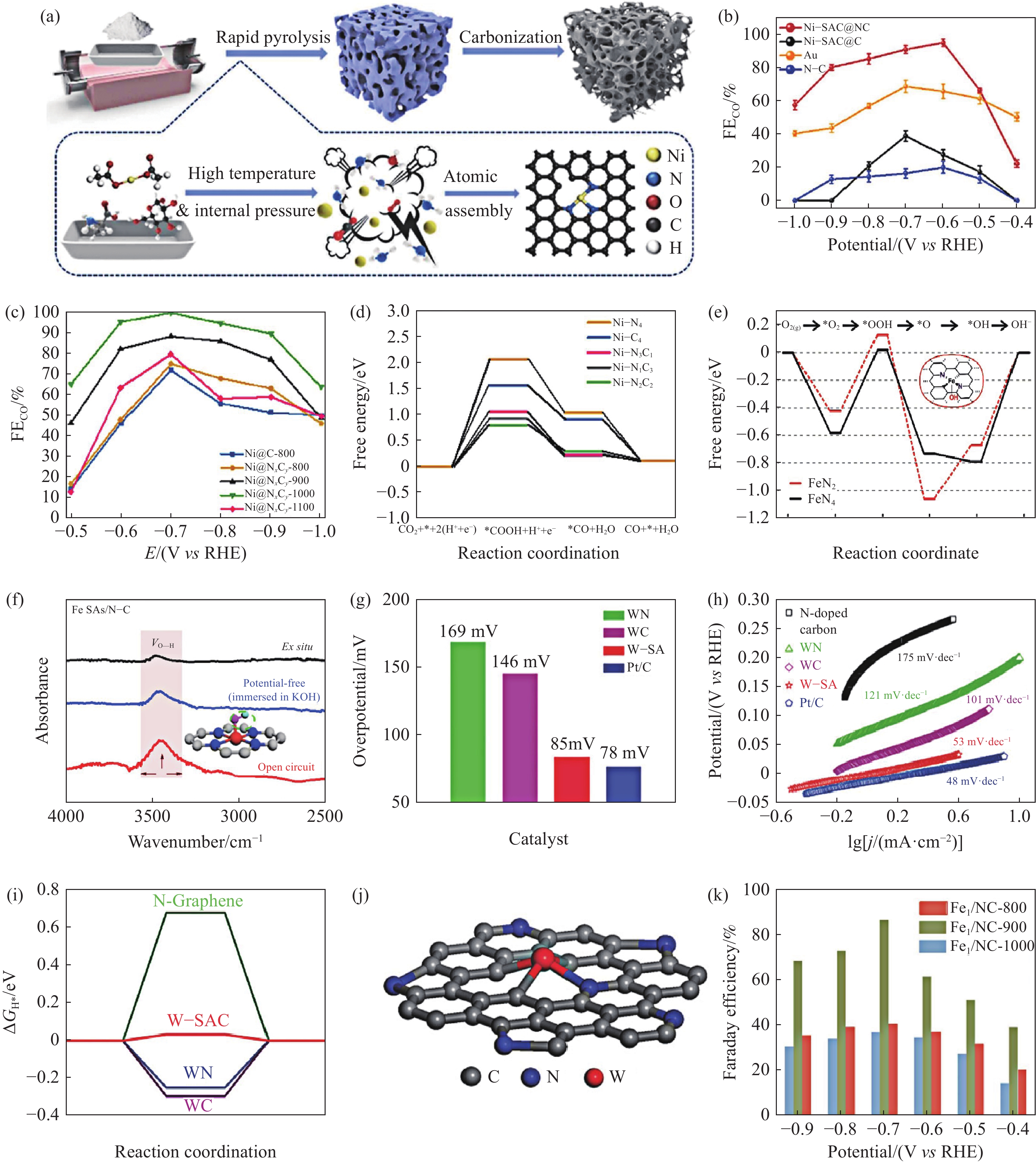
具有不对称配位结构的原子构型早前被Li和同事报道并用于CO2RR. 不对称Ni—N3Cl结构在电催化CO2RR方面取得了比Ni纳米粒子更大的突破. 此外,Ni—N3也被证明具有出色的CO2RR性能,在‒0.7 V至‒0.9 V条件下,FECO超过90%. 如图5(a)所示,Guo等[93]采用高效快速热解法制备了氮掺杂碳负载Ni SACs(Ni‒SAC@NCs),其结构为缺陷型Ni—N3. Ni‒SAC@NCs在‒0.6~‒0.9 V vs RHE时,法拉第效率保持在80%以上,在‒0.6 V vs RHE时,CO的法拉第效率最高达到95%(图5(b)). DFT结果表明缺陷型的Ni—N3结构降低了COOH*的形成势垒,进而加快了CO2反应进程. Yang等[94]通过控制热解过程中的温度,成功地调制备出NiN2C2构型的催化剂. 在‒0.7 V vs RHE的电位下实现了98.7%的CO法拉第效率(图5(c)). DFT计算证实Ni—N2C2具有最佳的催化性能(图5(d)). 活性位点可以产生更多未占据的三维轨道,从而大大降低了速率决定步骤的自由能,并表现出较高的CO2RR活性单个原子的化学状态.
![]() 图 5 (a) Ni SAC@NC的制备示意图[93]; (b) Ni SAC@NC在不同阴极电位下的法拉第效率[93]; (c) 不同温度下Ni@NxCy在不同阴极电位下的法拉第效率[94]; (d) Ni@NxCy位点上CO2RR到CO的吉布斯自由能图[94]; (e) FeN2和FeN4的ORR吉布斯自由能图[96]; (f) Fe SAC/N‒C的红外光谱图[97]; (g) W‒SA和对比催化剂在10 mA·cm‒2下的过电势[100]; (h) W‒SA和对比催化剂的Tafel[100]; (i) W‒SAC和对比催化剂对氢吸附的吉布斯自由能(ΔGH*)[100]; (j) W‒SAC原子结构模型[100]; (k) Fe1/NC-
图 5 (a) Ni SAC@NC的制备示意图[93]; (b) Ni SAC@NC在不同阴极电位下的法拉第效率[93]; (c) 不同温度下Ni@NxCy在不同阴极电位下的法拉第效率[94]; (d) Ni@NxCy位点上CO2RR到CO的吉布斯自由能图[94]; (e) FeN2和FeN4的ORR吉布斯自由能图[96]; (f) Fe SAC/N‒C的红外光谱图[97]; (g) W‒SA和对比催化剂在10 mA·cm‒2下的过电势[100]; (h) W‒SA和对比催化剂的Tafel[100]; (i) W‒SAC和对比催化剂对氢吸附的吉布斯自由能(ΔGH*)[100]; (j) W‒SAC原子结构模型[100]; (k) Fe1/NC-1000 ,Fe1/NC-900和Fe1/NC-800的FE[101]Figure 5. (a) Schematic illustration of Ni SAC@NC preparation[93]; (b) Faraday efficiency of Ni SAC@NC at different cathode potentials[93]; (c) Faraday efficiency of Ni@NxCy at different cathode potentials at different temperatures[94]; (d) Gibbs free energy diagram of CO2RR to CO at Ni@NxCy[94]; (e) ORR Gibbs free energy graphs for FeN2 and FeN4[96]; (f) infrared spectra of Fe SAC/N‒C[97]; (g) W‒SA and the overpotential of the catalyst compared at 10 mA·cm‒2[100]; (h) W‒SA and Tafel of the contrast catalyst[100]; (i) Gibbs free energy (ΔGH*) of the catalyst for hydrogen adsorption compared with W‒SAC[100]; (j) W‒SAC model of atomic structure[100]; (k) Fe1/NC-1000 , Fe1/NC-900, and Fe1/NC-800 FE[101]近年来,具有非对称配位的Fe SAC的合理设计也在如火如荼地进行. Li等[95]以活性炭黑为载体与Fe3+(Fe3+‒ACB)、尿素和PPh3的混合物在氩气气氛中热解制备了结构为Fe—N3P构型的Fe—N/P—C催化剂. Fe—N/P—C催化剂在电化学还原CO2为CO方面表现出色,法拉第效率高达98%,在‒0.34 V的低过电位下,质量归一化转换频率高达508.8 h−1. 基于非原位X射线吸收光谱测量和DFT计算结果发现,Fe SACs催化剂中P的调谐降低了Fe中心的氧化态,降低了*CO中间体形成的自由能垒,从而保持了Fe SACs催化剂的电催化活性和稳定性. Shen等[96]以掺氮有序介孔碳(NOMC)为载体制备了Fe−N2/NOMC SACs. 在该体系中,Fe—N2与中间产物*O2和*OH的结合能较低(图5(e)). 此外,高度有序的介孔结构加速了O2和电解质的传质. 因此,在碱性介质中,Fe−N2/NOMC催化剂的四电子还原活性和对O2的选择性均高于商业Pt/C催化剂. Zhou和他的同事[97]对Fe—N2活性位点提出了另一种新的观点. 发现在原子水平上,氢氧根可以以不饱和配位结构与铁原子偶联(图5(f)). 形成的OH—Fe—N2活性位点能促进O2的吸附,在低驱动电位下加速中间*OOH的形成,提高其ORR性能. 此外,Liu和他的同事[98]结合自组装和高温金属蚀刻工艺,将Fe—N3位均匀分散在多尺寸多孔碳框架上,记为Fe−N3/PCF. DFT计算结果表明,与分散在常规载体上的Fe—N4位相比,边缘支撑的Fe—N3位的本能谱活性显著增强,这极大地促进了ORR过程.
除了Fe、Ni外,Cu基SACs在电催化领域也占有非常重要的地位. 这归因于Cu具有独特的电子结构(3d轨道中没有价电子),使得Cu具有不同于其他元素的物理和化学性质. Han等[99]探索了不对称Cu—N2C2活性位点在ORR中的性能,并观察了反应过程中催化剂的动态变化. 事实上,Cu—N2C2的几何畸变是由新配位的含O物质刺激引起的. 这种活性位点和载体可以强化新的Cu—O键,削弱原有的Cu—N/Cu—C键,调节吸附剂的电子转移状态. 结果,大部分电子被转移到O2分子上,使ORR活性提高了6.59倍,除了M—N2和M—N3结构外,不对称的M—N1结构也被研究出具有高效的电催化性能. Chen等[100]通过热解将WCl5包封在UiO‒66‒NH2中,得到了碱性HER性能优异的W1—N1C3活性位点. 对于HER而言,如果催化剂表面吸附氢的吉布斯自由能(DGH*)较低,则其HER活性较高. DFT结果表明(图5(i)),W1—N1C3的DGH*值(0.033 eV)小于W—N2C2‒a、W—N2C2‒b、W—N3C1等结构. 如图5(g)和5(h)所示,在电流密度为10 mA·cm‒2时,过电位为85 mV,塔菲尔斜率低至53 mV·dec‒1.
NO3RR作为一种新兴的电化学合成氨的新技术,由于其能将水环境污染物——硝酸盐转化为有价值和可回收的NH3,且反应条件温和,在近两年受到了广泛的关注. 其中,Cu和Fe因其低廉的价格、优异的给电子能力和较高的活性而被认为是较好的NO3RR非贵金属催化剂. 选择Cu和Fe是因为它们的电催化活性优于其他过渡金属. Liu[101]应用常规的热解策略设计合成出Fe—N3构型的SACs催化剂(Fe‒NC-900). 如图5(k)所示,与FeN4相比,Fe‒NC-900催化剂在‒0.7 V vs RHE中表现出86.7%的法拉第效率,在‒0.9 V vs RHE中NH3产率约为18.8 mg·h−1·mg−1,在20 h的稳定性测试后依然保持了优异的法拉第效率和NH3产率. 电化学衰减全反射表面增强红外吸收光谱(EC‒ATR‒SEIRAS)和DFT模拟表明,Fe1/NC-900在NO3RR中表现出较高的活性,这是由于Fe—N3活性位点的孤对电子较多,且四面体顶端的Fe位点更有利于硝酸盐吸附,增强电荷转移,降低RDS的能垒. Cheng等[102]设计了打破对称性的Cu单原子催化剂. 在运行超过2000 h后仍保持有催化活性,在366 mA·cm‒2的工业水平电流密度下,氨的平均产率为27.84 mg·h‒1·cm‒2,在催化活性和长期稳定性之间取得了良好的平衡. 通过将一个Cu原子嵌入石墨烯纳米片中实现配位对称性破坏,其中两个N和两个O原子呈顺式构型,有效地降低了配位对称性,使活性位点更具极性,在电催化剂表面附近积累更多的NO3‒. 此外,与其他催化剂产生的σ配合物相比,顺式配位分裂了Cu 3d轨道,产生了与关键中间体*ONH的π配合物匹配的轨道对称性并降低了能垒.
2.3 M—M
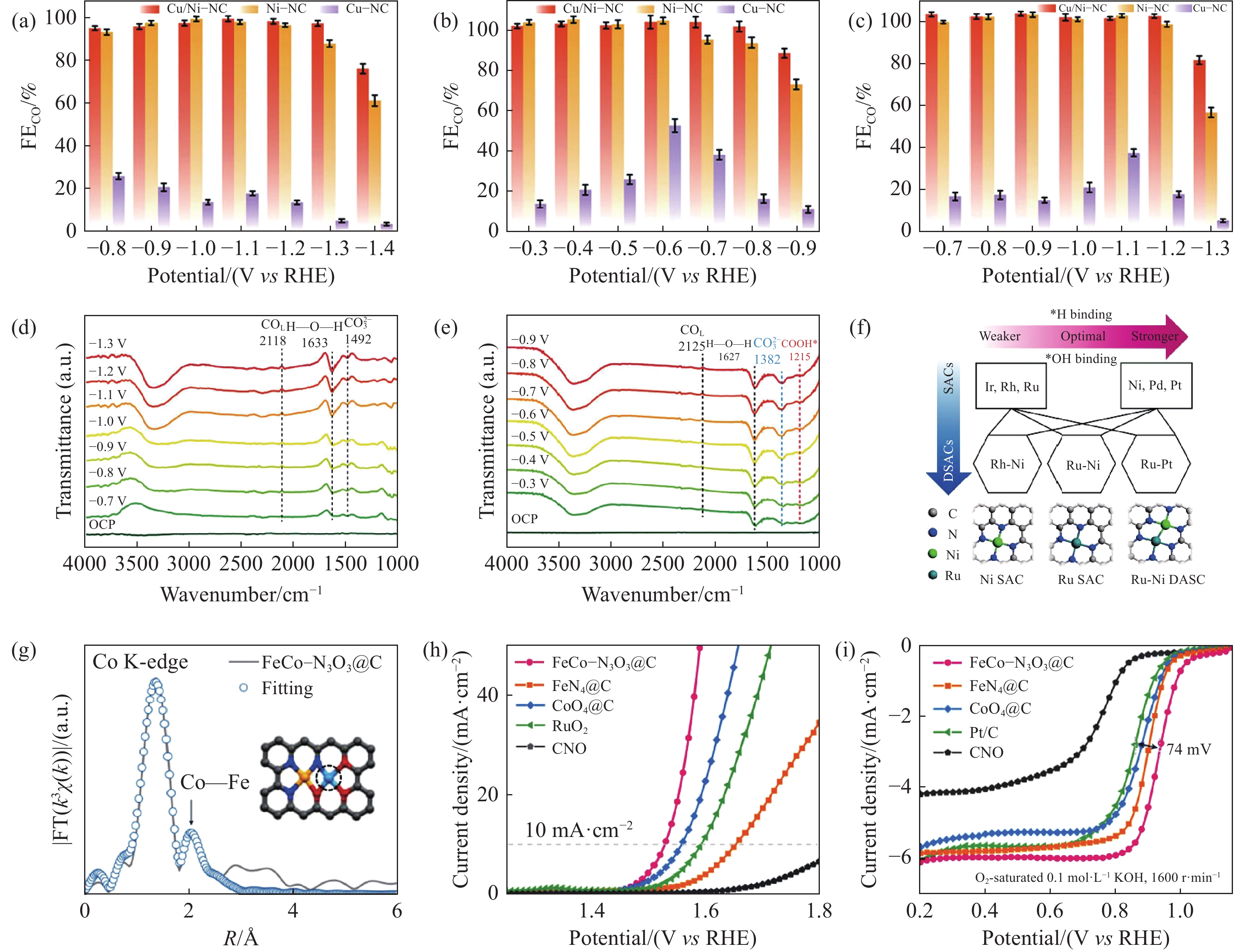
当前非对称结构的双金属位结构有M1—Nx和M2—Nx,M1M2—Nx及M1—M2,在电催化领域得到了广泛的研究. Zhang等[103]在金属有机骨架的帮助下,成功合成了一种配位良好的Ni/Cu DACs. 在全pH条件下(图6(a)~6(c)),Ni/Cu DACs均表现出宽电势范围内的高FECO. 尤其在酸性条件下,在流动池中经过25 h的耐久性测试后,仍有较好的CO2RR性能. 与Ni—NC样品的ATR‒SEIRAS结果相比较(图6(d)和6(e)),在反应过程中,CO2吸附在Cu/Ni—NC上,第一个电子转移形成*CO2‒中间体,然后形成的*CO2‒质子化形成*COOH,其中H+来自H2O(中性和碱性)或大量的电解质(酸性). *COOH随后脱水后在表面生成*CO中间体,最后*CO解吸形成目标产物. 这证明了相邻Ni和Cu活性位点的协同作用,从而获得显著的催化性能. Yun和他的同事[104]合成出的相同金属配位结构的Cu—Fe—N6—C催化剂,在CO2RR中同样表现出优异的FECO(在‒0.7 V时98%). 除上述金属配位结构相同的催化剂,Jiao等[105]采用牺牲模板法和双溶剂浸渍法制备了TeN2—CuN3 DACs. DACs表现出高FECO(>90%)的宽电位范围和良好的稳定性. Operando衰减全反射表面增强红外吸收光谱(ATR‒SEIRAS)发现,在不对称的TeN2—CuN3中,Te中心激活CO2,Cu中心帮助解离H2O,从而通过协同机制促进CO2RR过程. 研究发现,金属原子的配位数越低,催化性能越好. Liu等[106]通过溶解碳化法构建出CuSn/NPC DACs,其结构为CuN2SnN2. 与CuN3和SnN3相比,CuSn/NPC DACs催化剂优异的CO法拉第效率,且在24 h内稳定性保持良好. 同样的,Han等[107]采用相同的方法设计合成出RuNi DACs(图6(f)). EXAFS拟合结果表明DACs结构为RuN3NiN3. 与RuN3和NiN3相比,该催化剂在碱性电解质中表现出优异的氢氧化反应(HOR)活性,在过电位为50 mV时,其质量比动力学电流密度为(132.6±3.3) mA·mg−1(相对于RuNi质量) ,交换电流密度为(2.69±0.06) mA·cm−2. Guo等[108]利用级联热解法合成了负载缺陷富氮多孔碳的异核Ni—Ag双原子催化剂Ni—Ag/PC—N. XANES和WT结果证实了N3—Ni—Ag—N3结构的存在. 与Ag/PC—N、Ni/PC—N和无金属PCN相比,Ni—Ag/PC—N具有更好的CO选择性和更高的CO2RR电流密度. 这是由于配位的Ni原子降低了Ni—Ag/PC—N表面形成*COOH中间体的能垒,而相邻的Ag原子则减轻了Ni原子位点上*CO的强亲和力引起的催化剂中毒,进而提高了CO2RR性能.
![]() 图 6 (a) 酸性电解液[103]; (b) 碱性电解液和(c)中性电解液中Cu/Ni‒NC、Ni‒NC和Cu‒NC上CO的FEs[103]; (d) 酸性介质和 (e) 中性介质中Cu/Ni‒NC的原位ATR‒SEIRAS测量[103]; (f) 双原子组合筛选[107]; (g) FeCo‒N3O3@C中Co的K-edge XANES光谱及其理论光谱,插图用于模拟XANES光谱的结构(灰色、蓝色、红色、黄色和青色球分别代表碳、氮、氧、铁和钴原子)[110]; (h) 0.1 mol·L‒1 KOH下OER中FeCo‒N3O3@C、FeN4@C、CoO4@C、RuO2和CNO的LSV曲线[110]; (i) 在
图 6 (a) 酸性电解液[103]; (b) 碱性电解液和(c)中性电解液中Cu/Ni‒NC、Ni‒NC和Cu‒NC上CO的FEs[103]; (d) 酸性介质和 (e) 中性介质中Cu/Ni‒NC的原位ATR‒SEIRAS测量[103]; (f) 双原子组合筛选[107]; (g) FeCo‒N3O3@C中Co的K-edge XANES光谱及其理论光谱,插图用于模拟XANES光谱的结构(灰色、蓝色、红色、黄色和青色球分别代表碳、氮、氧、铁和钴原子)[110]; (h) 0.1 mol·L‒1 KOH下OER中FeCo‒N3O3@C、FeN4@C、CoO4@C、RuO2和CNO的LSV曲线[110]; (i) 在1600 r·min–1的0.1 mol·L‒1 KOH O2饱和溶液中,ORR中FeCo‒N3O3@C、FeN4@C、CoO4@C、Pt/C和CNO的LSV曲线[110]Figure 6. (a) Acid electrolyte[103]; (b) FEs of CO on Cu/ Ni‒NC, Ni‒NC, and Cu‒NC in alkaline electrolyte and (c) neutral electrolyte[103]; in-situ ATR‒SEIRAS measurements of Cu/Ni‒NC in (d) acidic media and (e) neutral media[103]; (f) diatomic combination screening[107]; (g) K-side XANES spectra and theoretical spectra of Co in FeCo‒N3O3@C, with inset simulating the structure of the XANES spectrum (grey, blue, red, yellow, and cyan spheres representing carbon, nitrogen, oxygen, iron, and cobalt atoms, respectively)[110]; (h) LSV curves of FeCo‒N3O3@C, FeN4@C, CoO4@C, RuO2, and CNO in OER at 0.1 mol·L‒1 KOH[110]; (i) LSV curves of FeCo‒N3O3@C, FeN4@C, CoO4@C, Pt/C, and CNO in ORR in the 0.1 mol·L‒1 KOH O2 saturated solution at1600 r·min–1[110]ORR是燃料电池和金属空气电池等新型能量存储和转换系统中必不可少的反应之一. 然而,ORR的电子传递多步且速度慢,传质效率低,导致其动力学速度慢,这极大地阻碍了其在实际器件中的应用. 因此,提高ORR催化剂的效率和耐久性,能够直接提升这些能量转换装置的性能和稳定性,从而推动其在能源领域的广泛应用. Yang等[109]以双氰胺为C源和N源,酞菁铁和硝酸锰为金属前驱体,通过预聚合和热解制备了Fe—N4和Mn—N4活性位点相邻的Fe、Mn/N—C SACs. 与Fe/NC对比,Mn—N基团的引入合理调节了体系的自旋态密度,有效激活了Fe3+位点,使催化剂在酸性(E1/2为0.804 V)和碱性(E1/2为0.928 V)条件下均表现出优异的ORR活性和稳定性. 另一方面,Tang等[110]以简单的“一锅法”制备了桥连N和O原子的Fe—Co DACs(FeCo‒N3O3@C),如图6(g)所示. 该催化剂是ORR(E1/2= 0.936 V)和OER(E= 1.528 V, 10 mA·cm−2)中稳定高效的双功能催化剂(图6(h)和6(i)). 在组成的可充电水性锌‒空气电池中能够稳定200 h. 同样具有双功能电催化作用的IrCo—N5活性位点由Xiao等[111]提出. FT‒EXAFS光谱显示样品中存在Ir—N,Co—N和Ir—Co键,进一步证实了不对称的活性结. 用10 mA·cm‒2的OER和ORR的E1/2之间的电位差来评估双功能催化活性(DE). 研究发现,IrCo—N—C具有最好的DE,低至0.649 V,甚至优于目前最先进的Pt/C+Ir/C对(0.706 V). 理论计算表明,优异的ORR和OER活性是由于Ir原子的加入. 它改变了Co原子本身的d轨道能级,增强了Co位的自旋极化态,改善了氧的吸附态.
总的来说,相较于SACs,双原子催化剂通过引入两个相互作用的原子作为活性位点,具有更多的活性位点,在复杂的催化反应中,双金属的协同作用打破了单核位点的吸附关联限制,提高了反应中间体的吸附脱附,降低了反应能垒,从而优化了反应路径和提高了催化效率.
3. 总结与展望
本文总结了当前非对称配位结构SACs的最新研究进展,包括它们的通用合成方法及在能源催化中的应用,为SACs结构调控提供参考. 此外,本文还探讨了不同结构在催化反应中的优势及机制,证明了调控SACs配位环境策略的能够显著提高催化活性和选择性,且在未来研究发展中具有很大的潜力. 虽然SACs的配位微环境调控策略已逐步受到研究人员的重视,同时也在多种催化领域已经有广泛的应用研究. 然而,对于各种催化反应中的产物选择性、反应路径优化等还有很大进步空间. 因此,针对非对称配位结构SACs的未来发展还需要关注以下问题:
(1) 制备技术与工艺创新:目前,非对称配位SACs的制备仍面临一定的挑战,如制备成本高、产量有限等. 未来需要开发更高效、更经济的制备方法和工艺,以实现催化剂的规模化生产. 另一方面,探索新的制备技术和工艺,如静电吸附法、共沉淀法等的改进和优化,以及新型脉冲放电策略、热置换策略等的应用,以制备出性能更优、稳定性更好的非对称配位SACs. 重要的是,采用原位电镜、原位XRD等原位表征技术,检测非对称配位结构的SACs合成动态演变过程,为非对称配位结构的SACs的普适性方法做铺垫.
(2) 性能优化与提升:到目前为止,当前SACs的稳定性仍有较大的问题,对于实际的工业化应用存在阻碍. 通过选择合适的配体对于稳定SACs的配位环境至关重要,不同的配体对金属原子的配位能力和稳定性不同.
(3) 动态反应过程中催化机理探索:当前,已有大量的理论计算证明了不同构型的SACs的反应过程,实际过程中SACs的反应步骤是复杂的,未来应通过先进的原位表征手段如原位电镜、原位XRD、原位拉曼光谱等手段与理论计算相结合,阐明催化剂的反应路径.
(4) 更多领域的应用. 迄今为止,非对称配位结构SACs主要集中在ORR,CO2RR催化中,在电解水、NO3RR及有机分子转化中的研究相对很少. 未来非对称配位结构SACs扩展到这些领域,将会极大的发挥其潜力.
-
![]()
图 1 轴向配位的SACs合成. (a) Ni—N4—O/C的合成工艺说明[45]; (b) Ni—N4—O/C在R空间中的EXAFS拟合曲线,插图为Ni—N4—O/C相应的示意图模型[45]; (c) P‒CoPc@CNT合成示意图[39]; (d) 电沉积法合成Cl—Pt/LDH催化剂及可逆轴向配体交换过程示意图(原子由球体表示:Pt (蓝色),Ni (橄榄色),Fe (黄色),O (红色),Cl (绿色)和H (灰色))[49]
Figure 1. Synthesis of SACs with axial coordination: (a) schematic illustration of the synthesis process of Ni—N4—O /C[45]; (b) Ni—N4—O/C EXAFS fitting curve in R-space, with the inset showing the corresponding schematic model of Ni—N4—O /C[45]; (c) P‒CoPc@CNT composite diagram[39]; (d) schematic of synthesis of Cl—Pt/LDH catalyst by electrodeposition and reversible axial ligand exchange process (Atoms are represented by spheres: Pt (blue), Ni (olive), Fe (yellow), O (red), Cl (green), and H (gray)) [49]
![]()
图 2 杂原子掺杂的SACs合成. (a) Ni—NC/NHCSs-Y合成示意图[57]; (b) Ni–NC/NHCSs-600和Ni箔的Ni K-edge XANES光谱,插图为Ni–NC/NHCSs-600的扩展前边缘区域(峰A)[57]; (c) k3加权傅立叶变换EXAFS光谱(FTEXAFS)[57]; (d) Ni—N—C、Ni箔和NiPc的Ni K-edge XANES光谱[61]; (e‒g) Fe—N4,FeS1N3和FeB1N3在R空间中的EXAFS拟合曲线,插图为相应的示意图模型[63]
Figure 2. Synthesis of SACs doped with heteroatoms: (a) schematic illustration of Ni—NC/NHCSs-Y synthesis[57]; (b) Ni K-edge XANES spectra of Ni–NC/NHCSs-600 and Ni foils, with the inset showing extended front edge region of Ni–NC/NHCSs-600 (Peak A)[57]; (c) k3 weighted Fourier transform EXAFS spectrum (FTEXAFS)[57]; (d) Ni K-Edge XANES spectra of Ni—N—C, Ni foil, and NiPc[61]; (e‒g) EXAFS fitted curves of Fe—N4, FeS1N3, and FeB1N3 in R-space, respectively. The insets depict the corresponding schematic models[63]
![]()
图 3 双金属位点的SACs合成. (a) Fe2NPC结构示意图[78]; (b) Fe2NPC的HAADF‒STEM图像[78]; (c) Fe2NPC的EXAFS R空间拟合曲线[78]; (d) 制备ZnRu‒、NiRu‒、ZnCu‒、CoCu‒、NiCu‒和BiCu‒PCN异核DACs的导航定位策略示意图[82]; (e) Ni/Cu—N—C制备原理[86]
Figure 3. Synthesis of bimetallic atomic sites: (a) Fe2NPC structure diagram[78]; (b) HAADF‒STEM images of Fe2NPC[78]; (c) Fe2NPC EXAFS R-space fitting curve[78]; (d) preparation of ZnRu‒, NiRu‒, ZnCu‒, CoCu‒, NiCu‒, and BiCu‒PCN heteronuclear DACs navigation strategy diagram[82]; (e) Ni/Cu—N—C preparation principle[86]
![]()
图 4 (a) SnPc/CNT−OH的制备示意图[88]; (b) Ⅰ:SnPc/CNT−OH、Ⅱ:SnPc/CNT−NH2和Ⅲ:SnPc/CNT在不同阴极电位下的法拉第效率[88]; (c) O—Sn—N4和Sn—N4位点上CO2RR到HCOOH的吉布斯自由能图[88]; (d) 0.1 M KOH下FePc、FePc/CNT‒NH2和Pt/C的LSV曲线[14]; (e和f) HER极化曲线[92]
Figure 4. (a) Schematic illustration of the preparation of SnPc/CNT−OH[88]; (b) Faraday efficiencies of Ⅰ: SnPc/CNT−OH, Ⅱ: SnPc/CNT−NH2, and Ⅲ: SnPc/CNT at different cathode potentials[88]; (c) Gibbs free energy graphs of CO2RR to HCOOH at O—Sn—N4 and Sn—N4 sites[88]; (d) LSV curves of FePc, FePc/CNT‒NH2, and Pt/C at 0.1 M KOH[14]; (e and f) HER polarization curve[92]
![]()
图 5 (a) Ni SAC@NC的制备示意图[93]; (b) Ni SAC@NC在不同阴极电位下的法拉第效率[93]; (c) 不同温度下Ni@NxCy在不同阴极电位下的法拉第效率[94]; (d) Ni@NxCy位点上CO2RR到CO的吉布斯自由能图[94]; (e) FeN2和FeN4的ORR吉布斯自由能图[96]; (f) Fe SAC/N‒C的红外光谱图[97]; (g) W‒SA和对比催化剂在10 mA·cm‒2下的过电势[100]; (h) W‒SA和对比催化剂的Tafel[100]; (i) W‒SAC和对比催化剂对氢吸附的吉布斯自由能(ΔGH*)[100]; (j) W‒SAC原子结构模型[100]; (k) Fe1/NC-
1000 ,Fe1/NC-900和Fe1/NC-800的FE[101]Figure 5. (a) Schematic illustration of Ni SAC@NC preparation[93]; (b) Faraday efficiency of Ni SAC@NC at different cathode potentials[93]; (c) Faraday efficiency of Ni@NxCy at different cathode potentials at different temperatures[94]; (d) Gibbs free energy diagram of CO2RR to CO at Ni@NxCy[94]; (e) ORR Gibbs free energy graphs for FeN2 and FeN4[96]; (f) infrared spectra of Fe SAC/N‒C[97]; (g) W‒SA and the overpotential of the catalyst compared at 10 mA·cm‒2[100]; (h) W‒SA and Tafel of the contrast catalyst[100]; (i) Gibbs free energy (ΔGH*) of the catalyst for hydrogen adsorption compared with W‒SAC[100]; (j) W‒SAC model of atomic structure[100]; (k) Fe1/NC-
1000 , Fe1/NC-900, and Fe1/NC-800 FE[101]![]()
图 6 (a) 酸性电解液[103]; (b) 碱性电解液和(c)中性电解液中Cu/Ni‒NC、Ni‒NC和Cu‒NC上CO的FEs[103]; (d) 酸性介质和 (e) 中性介质中Cu/Ni‒NC的原位ATR‒SEIRAS测量[103]; (f) 双原子组合筛选[107]; (g) FeCo‒N3O3@C中Co的K-edge XANES光谱及其理论光谱,插图用于模拟XANES光谱的结构(灰色、蓝色、红色、黄色和青色球分别代表碳、氮、氧、铁和钴原子)[110]; (h) 0.1 mol·L‒1 KOH下OER中FeCo‒N3O3@C、FeN4@C、CoO4@C、RuO2和CNO的LSV曲线[110]; (i) 在
1600 r·min–1的0.1 mol·L‒1 KOH O2饱和溶液中,ORR中FeCo‒N3O3@C、FeN4@C、CoO4@C、Pt/C和CNO的LSV曲线[110]Figure 6. (a) Acid electrolyte[103]; (b) FEs of CO on Cu/ Ni‒NC, Ni‒NC, and Cu‒NC in alkaline electrolyte and (c) neutral electrolyte[103]; in-situ ATR‒SEIRAS measurements of Cu/Ni‒NC in (d) acidic media and (e) neutral media[103]; (f) diatomic combination screening[107]; (g) K-side XANES spectra and theoretical spectra of Co in FeCo‒N3O3@C, with inset simulating the structure of the XANES spectrum (grey, blue, red, yellow, and cyan spheres representing carbon, nitrogen, oxygen, iron, and cobalt atoms, respectively)[110]; (h) LSV curves of FeCo‒N3O3@C, FeN4@C, CoO4@C, RuO2, and CNO in OER at 0.1 mol·L‒1 KOH[110]; (i) LSV curves of FeCo‒N3O3@C, FeN4@C, CoO4@C, Pt/C, and CNO in ORR in the 0.1 mol·L‒1 KOH O2 saturated solution at
1600 r·min–1[110] -
[1] Zhang Y, Jiang H, Zhang C X, et al. High-efficiency oxygen reduction by late transition metal oxides to produce H2O2. J Mater Chem A, 2024, 12(10): 6123 doi: 10.1039/D3TA06995H
[2] Zhang W D, Dong H L, Zhou L, et al. Fe single-atom catalysts with pre-organized coordination structure for efficient electrochemical nitrate reduction to ammonia. Appl Catal B Environ, 2022, 317: 121750 doi: 10.1016/j.apcatb.2022.121750
[3] Hu J, Guo T Q, Zhong X Y, et al. In situ reconstruction of high-entropy heterostructure catalysts for stable oxygen evolution electrocatalysis under industrial conditions. Adv Mater, 2024, 36(14): 2310918 doi: 10.1002/adma.202310918
[4] Lu R H, Quan C X, Zhang C Y, et al. Establishing a theoretical insight for penta-coordinated iron-nitrogen-carbon catalysts toward oxygen reaction. Nano Res, 2022, 15(7): 6067 doi: 10.1007/s12274-022-4318-2
[5] 夏伟, 黎小辉, 白婷, 等. 甲烷干重整与二氧化碳甲烷化的工艺耦合研究. 工程科学学报, 2024, 46(11):2110 Xia W, Li X H, Bai T, et al. Study on process coupling between dry reforming of methane and methanation of carbon dioxide. Chin J Eng, 2024, 46(11): 2110
[6] 习爽, 程溪明, 高兴伟, 等. 面向锌离子电池的二氧化锰柔性电极制备及电化学性能. 工程科学学报, 2024, 46(11):2036 Xi S, Cheng X M, Gao X W, et al. Preparation and electrochemical properties of manganese dioxide flexible electrodes for zinc-ion batteries. Chin J Eng, 2024, 46(11): 2036
[7] Qiao B T, Wang A Q, Yang X F, et al. Single-atom catalysis of CO oxidation using Pt1/FeO x. Nat Chem, 2011, 3(8): 634 doi: 10.1038/nchem.1095
[8] 胡觉, 姚耀春, 张呈旭. 新能源材料与器件概论. 北京:冶金工业出版社, 2022 Hu J, Yao Y C, Zhang C X. Introduction to New Energy Materials and Devices. Beijing: Metallurgical Industry Press, 2022
[9] He W Y, Wu J H, Liu J L, et al. Single-atom nanozymes for catalytic therapy: Recent advances and challenges. Adv Funct Mater, 2024, 34(16): 2312116 doi: 10.1002/adfm.202312116
[10] Zhou R, Gu S N, Guo M, et al. Progresses and prospects of asymmetrically coordinated single atom catalysts for lithium−sulfur batteries. Energy Environ Mater, 2024, 7(4): e12703 doi: 10.1002/eem2.12703
[11] 胡觉. 电解水制氢催化剂. 北京:冶金工业出版社, 2022 Hu J. Electrocatalysts for Efficient Water Splitting. Beijing: Metallurgical Industry Press, 2022
[12] Wang Z D, Yuan X Y, Guo H, et al. Rational design principles of single-atom catalysts for hydrogen production and hydrogenation. Energy Environ Sci, 2024, 17(21): 8019 doi: 10.1039/D4EE02763A
[13] 司庆宇, 樊星, 张玮航, 等. Fe和Cu分子筛催化剂同时催化NO x还原和N2O分解. 中国环境科学, 2024, 44(9):4826 doi: 10.3969/j.issn.1000-6923.2024.09.009 Si Q Y, Fan X, Zhang W H, et al. Simultaneous reduction of NO x and decomposition of N2O over Fe and Cu zeolite catalysts. China Environ Sci, 2024, 44(9): 4826 doi: 10.3969/j.issn.1000-6923.2024.09.009
[14] Zhang W L, Meeus E J, Wang L, et al. Boosting electrochemical oxygen reduction performance of iron phthalocyanine through axial coordination sphere interaction. Chem Sus Chem, 2022, 15(3): e202102379 doi: 10.1002/cssc.202102379
[15] Yang H B, Hung S F, Liu S, et al. Atomically dispersed Ni(i) as the active site for electrochemical CO2 reduction. Nat Energy, 2018, 3(2): 140 doi: 10.1038/s41560-017-0078-8
[16] Lu Z Y, Wang B, Hu Y F, et al. An isolated zinc–cobalt atomic pair for highly active and durable oxygen reduction. Angew Chem Int Ed, 2019, 58(9): 2622 doi: 10.1002/anie.201810175
[17] Zhu X F, Zhang D T, Chen C J, et al. Harnessing the interplay of Fe‒Ni atom pairs embedded in nitrogen-doped carbon for bifunctional oxygen electrocatalysis. Nano Energy, 2020, 71: 104597 doi: 10.1016/j.nanoen.2020.104597
[18] Wang Y L, Yin H B, Dong F, et al. N-coordinated Cu‒Ni dual-single-atom catalyst for highly selective electrocatalytic reduction of nitrate to ammonia. Small, 2023, 19(20): 2207695 doi: 10.1002/smll.202207695
[19] Wang X, Yi Z Y, Wang Y Q, et al. Molecular evidence for the axial coordination effect of atomic iodine on Fe-N4 sites in oxygen reduction reaction. Angew Chem Int Ed, 2025, 64(1): e202413673 doi: 10.1002/anie.202413673
[20] Sun J, Yang H X, Gao W Q, et al. Diatomic Pd−Cu metal-phosphorus sites for complete N≡N bond formation in photoelectrochemical nitrate reduction. Angew Chem Int Ed, 2022, 61(45): e202211373 doi: 10.1002/anie.202211373
[21] Jasinski R. A new fuel cell cathode catalyst. Nature, 1964, 201(4925): 1212 doi: 10.1038/2011212a0
[22] Sun Z Y, Li C, Wei Z H, et al. Sulfur-bridged asymmetric CuNi bimetallic atom sites for CO2 reduction with high efficiency. Adv Mater, 2024, 36(33): 2404665 doi: 10.1002/adma.202404665
[23] 张景涛, 王奎, 刘乐, 等. MOF衍生的Fe‒N‒C电催化剂, 用于高效氧还原反应. 功能材料, 2024, 55(8):8103 doi: 10.3969/j.issn.1001-9731.2024.08.014 Zhang J T, Wang K, Liu (L /Y), et al. MOF-derived Fe‒N‒C electrocatalyst for efficient oxygen reduction reactions. J Funct Mater, 2024, 55(8): 8103 doi: 10.3969/j.issn.1001-9731.2024.08.014
[24] Magnier L, Cossard G, Martin V, et al. Fe‒Ni-based alloys as highly active and low-cost oxygen evolution reaction catalyst in alkaline media. Nat Mater, 2024, 23(2): 252 doi: 10.1038/s41563-023-01744-5
[25] Yang B C, Huang J, Tong J, et al. Microwave synthesis of Fe‒Cu diatomic active center MOF: Synergistic cyclic catalysis of persulfate for degrading norfloxacin. Environ Sci: Nano, 2023, 10(10): 2778 doi: 10.1039/D3EN00340J
[26] 陈永杰, 邢小芳, 王阳, 等. Fe基CO2加氢制C2+醇催化剂研究进展. 燃料化学学报, 2024, 52(11):1580 doi: 10.3724/2097-213X.2024.JFCT.0007 Chen Y J, Xing X F, Wang Y, et al. Advances in Fe-based catalysts for the hydrogenation of CO2 to C2+ alcohols. J Fuel Chem Technol, 2024, 52(11): 1580 doi: 10.3724/2097-213X.2024.JFCT.0007
[27] Su X, Yang X F, Huang Y Q, et al. Single-atom catalysis toward efficient CO2 conversion to CO and formate products. Acc Chem Res, 2019, 52(3): 656 doi: 10.1021/acs.accounts.8b00478
[28] Sun Z Y, Ma T, Tao H C, et al. Fundamentals and challenges of electrochemical CO2 reduction using two-dimensional materials. Chem, 2017, 3(4): 560 doi: 10.1016/j.chempr.2017.09.009
[29] Ju W, Bagger A, Wang X L, et al. Unraveling mechanistic reaction pathways of the electrochemical CO2 reduction on Fe‒N‒C single-site catalysts. ACS Energy Lett, 2019, 4(7): 1663 doi: 10.1021/acsenergylett.9b01049
[30] Li Z, Jiang J X, Liu X M, et al. Coupling atomically dispersed Fe‒N5 sites with defective N-doped carbon boosts CO2 electroreduction. Small, 2022, 18(38): 2203495 doi: 10.1002/smll.202203495
[31] Guan W Q, Shao H D, Zhang C X, et al. Strategies for the regulation of specific active sites in metal−nitrogen−carbon. Nano Energy, 2024, 120: 109149 doi: 10.1016/j.nanoen.2023.109149
[32] Wang Y Z, Yang T M, Fan X, et al. Anchoring Fe species on the highly curved surface of S and N Co-doped carbonaceous nanosprings for oxygen electrocatalysis and a flexible zinc-air battery. Angew Chem Int Ed, 2024, 63(7): e202313034 doi: 10.1002/anie.202313034
[33] Kang Y K, Li Y F, Liu Z P. Unveiling curvature effect on Fe atom embedded N-doped carbon nanotubes for electrocatalytic oxygen reduction reactions using hybrid quantum-mechanics/machine-learning potential. J Phys Chem C, 2024, 128(8): 3127 doi: 10.1021/acs.jpcc.3c08073
[34] Zhou W L, Su H, Cheng W R, et al. Regulating the scaling relationship for high catalytic kinetics and selectivity of the oxygen reduction reaction. Nat Commun, 2022, 13(1): 6414 doi: 10.1038/s41467-022-34169-w
[35] Tang S H, Xu L, Dong K J, et al. Curvature effect on graphene-based Co/Ni single-atom catalysts. Appl Surf Sci, 2023, 615: 156357 doi: 10.1016/j.apsusc.2023.156357
[36] Xu W Q, Song W Y, Kang Y K, et al. Axial ligand-engineered single-atom catalysts with boosted enzyme-like activity for sensitive immunoassay. Anal Chem, 2021, 93(37): 12758 doi: 10.1021/acs.analchem.1c02842
[37] Zhang H C, Cui P X, Xie D H, et al. Axial N ligand-modulated ultrahigh activity and selectivity hyperoxide activation over single-atoms nanozymes. Adv Sci, 2023, 10(3): 2205681 doi: 10.1002/advs.202205681
[38] Yan L N, Wang C, Wang Y S, et al. Optimizing the binding of the *OOH intermediate via axially coordinated Co‒N5 motif for efficient electrocatalytic H2O2 production. Appl Catal B Environ, 2023, 338: 123078 doi: 10.1016/j.apcatb.2023.123078
[39] Liu Y, Zhang S S, Jiao C, et al. Axial phosphate coordination in Co single atoms boosts electrochemical oxygen evolution. Adv Sci, 2023, 10(5): 2206107 doi: 10.1002/advs.202206107
[40] Hu L Y, Dai C L, Chen L W, et al. Metal-triazolate-framework-derived FeN4Cl1 single-atom catalysts with hierarchical porosity for the oxygen reduction reaction. Angew Chem Int Ed, 2021, 60(52): 27324 doi: 10.1002/anie.202113895
[41] Huang M, Deng B W, Zhao X L, et al. Template-sacrificing synthesis of well-defined asymmetrically coordinated single-atom catalysts for highly efficient CO2 electrocatalytic reduction. ACS Nano, 2022, 16(2): 2110 doi: 10.1021/acsnano.1c07746
[42] Li L B, Huang S H, Cao R, et al. Optimizing microenvironment of asymmetric N, S-coordinated single-atom Fe via axial fifth coordination toward efficient oxygen electroreduction. Small, 2022, 18(2): 2105387 doi: 10.1002/smll.202105387
[43] Wang J, Zheng M Y, Zhao X, et al. Structure-performance descriptors and the role of the axial oxygen atom on M‒N4‒C single-atom catalysts for electrochemical CO2 reduction. ACS Catal, 2022, 12(9): 5441 doi: 10.1021/acscatal.2c00429
[44] Zhao K M, Liu S Q, Li Y Y, et al. Insight into the mechanism of axial ligands regulating the catalytic activity of Fe‒N4 sites for oxygen reduction reaction. Adv Energy Mater, 2022, 12(11): 2103588 doi: 10.1002/aenm.202103588
[45] Li X F, Han S G, Wu W M, et al. Convergent paired electrosynthesis of dimethyl carbonate from carbon dioxide enabled by designing the superstructure of axial oxygen coordinated nickel single-atom catalysts. Energy Environ Sci, 2023, 16(2): 502 doi: 10.1039/D2EE03022E
[46] Wang Z D, Han Y, Li B, et al. Regulation of electrocatalytic behavior by axial oxygen enhances the catalytic activity of CoN4 sites for CO2 reduction. Small, 2023, 19(34): 2301797 doi: 10.1002/smll.202301797
[47] Wang X Y, Wang Y, Sang X H, et al. Dynamic activation of adsorbed intermediates via axial traction for the promoted electrochemical CO2 reduction. Angew Chem Int Ed, 2021, 60(8): 4192 doi: 10.1002/anie.202013427
[48] Hu C H, Zhang Y, Hu A Q, et al. Near- and long-range electronic modulation of single metal sites to boost CO2 electrocatalytic reduction. Adv Mater, 2023, 35(19): 2209298 doi: 10.1002/adma.202209298
[49] Zhang T Y, Jin J, Chen J M, et al. Pinpointing the axial ligand effect on platinum single-atom-catalyst towards efficient alkaline hydrogen evolution reaction. Nat Commun, 2022, 13(1): 6875 doi: 10.1038/s41467-022-34619-5
[50] Ma Y R, Zhang M L, Wu J, et al. The key effect of carboxyl group and CuN2O2 coordinate structure for Cu, N Co-doped carbon dots with peroxidase-like property. Small, 2023, 19(31): 2300883 doi: 10.1002/smll.202300883
[51] Shi H N, Liang Y, Hou J G, et al. Boosting solar-driven CO2 conversion to ethanol via single-atom catalyst with defected low-coordination Cu‒N2 motif. Angew Chem Int Ed, 2024, 63(31): e202404884 doi: 10.1002/anie.202404884
[52] Gong Y N, Jiao L, Qian Y Y, et al. Regulating the coordination environment of MOF-templated single-atom nickel electrocatalysts for boosting CO2 reduction. Angew Chem Int Ed, 2020, 59(7): 2705 doi: 10.1002/anie.201914977
[53] Fan Z Z, Luo R C, Zhang Y X, et al. Oxygen-bridged indium-nickel atomic pair as dual-metal active sites enabling synergistic electrocatalytic CO2 reduction. Angew Chem Int Ed, 2023, 62(7): e202216326 doi: 10.1002/anie.202216326
[54] Zhang N Q, Zhang X X, Kang Y K, et al. A supported Pd2 dual-atom site catalyst for efficient electrochemical CO2 reduction. Angew Chem Int Ed, 2021, 60(24): 13388 doi: 10.1002/anie.202101559
[55] Gong Q F, Wang Y J, Ren X Z, et al. Ultra-low-loaded Ni−Fe dimer anchored to nitrogen/oxygen sites for boosting electroreduction of carbon dioxide. ChemSusChem, 2021, 14(20): 4499 doi: 10.1002/cssc.202101302
[56] Li Y F, Chen C, Cao R, et al. Dual-atom Ag2/graphene catalyst for efficient electroreduction of CO2 to CO. Appl Catal B Environ, 2020, 268: 118747 doi: 10.1016/j.apcatb.2020.118747
[57] Gong S H, Wang W B, Lu R Q, et al. Mediating heterogenized nickel phthalocyanine into isolated Ni‒N3 moiety for improving activity and stability of electrocatalytic CO2 reduction. Appl Catal B Environ, 2022, 318: 121813 doi: 10.1016/j.apcatb.2022.121813
[58] Yan C C, Li H B, Ye Y F, et al. Coordinatively unsaturated nickel–nitrogen sites towards selective and high-rate CO2 electroreduction. Energy Environ Sci, 2018, 11(5): 1204 doi: 10.1039/C8EE00133B
[59] Wang X Q, Chen Z, Zhao X Y, et al. Regulation of coordination number over single Co sites: Triggering the efficient electroreduction of CO2. Angew Chem Int Ed, 2018, 57(7): 1944 doi: 10.1002/anie.201712451
[60] A. l. B. Delphine Cabaret, Ame´ lie Juhin, Mounir Arfaoui and Emilie Gaudry, First-principles calculations of X-ray absorption spectra at the K-edge of 3d transition metals: an electronic structure analysis of the pre-edge. Phys Chem Chem Phys, 2010, 12(21): 5619-5633
[61] Zhou Y Z, Zhou Q, Liu H J, et al. Asymmetric dinitrogen-coordinated nickel single-atomic sites for efficient CO2 electroreduction. Nat Commun, 2023, 14(1): 3776 doi: 10.1038/s41467-023-39505-2
[62] Zhang W, Liu D, Liu T, et al. Coordinately unsaturated nickel single atom electrocatalyst for efficient CO2 conversion. Nano Res, 2023, 16(8): 10873 doi: 10.1007/s12274-023-5949-7
[63] Jin Z Y, Jiao D X, Dong Y L, et al. Boosting electrocatalytic carbon dioxide reduction via self-relaxation of asymmetric coordination in Fe-based single atom catalyst. Angew Chem, 2024, 136(6): e202318246 doi: 10.1002/ange.202318246
[64] Yang X, Cheng J, Lv H K, et al. Sulfur-doped unsaturated Ni‒N3 coordination for efficient electroreduction of CO2. Chem Eng J, 2022, 450: 137950 doi: 10.1016/j.cej.2022.137950
[65] Xu J W, Zhang S B, Liu H J, et al. Breaking local charge symmetry of iron single atoms for efficient electrocatalytic nitrate reduction to ammonia. Angew Chem Int Ed, 2023, 62(39): e202308044 doi: 10.1002/anie.202308044
[66] Wan J W, Zhao Z H, Shang H S, et al. In situ phosphatizing of triphenylphosphine encapsulated within metal-organic frameworks to design atomic Co1–P1N3 interfacial structure for promoting catalytic performance. J Am Chem Soc, 2020, 142(18): 8431 doi: 10.1021/jacs.0c02229
[67] Shao X D, Gan R H, Rao Y, et al. Main Group SnN4O single sites with optimized charge distribution for boosting the oxygen reduction reaction. ACS Nano, 2024, 18(22): 14742 doi: 10.1021/acsnano.4c04112
[68] Li Y Z, Sun H, Ren L T, et al. Asymmetric coordination regulating D-orbital spin-electron filling in single-atom iron catalyst for efficient oxygen reduction. Angew Chem Int Ed, 2024, 63(28): e202405334 doi: 10.1002/anie.202405334
[69] Zhang F W, Zhang H, Jia Z H, et al. Nickel single atom density-dependent CO2 efficient electroreduction. Small, 2024, 20(16): 2308080 doi: 10.1002/smll.202308080
[70] Yu K, Sun K A, Cheong W C, et al. Oxalate-assisted synthesis of hollow carbon nanocage with Fe single atoms for electrochemical CO2 reduction. Small, 2023, 19(39): 2302611 doi: 10.1002/smll.202302611
[71] Bie Q Q, Yin H B, Wang Y L, et al. Electrocatalytic reduction of CO2 with enhanced C2 liquid products activity by the synergistic effect of Cu single atoms and oxygen vacancies. Chin J Catal, 2024, 57: 96 doi: 10.1016/S1872-2067(23)64587-5
[72] Zhang Y X, Zhang S B, Huang H L, et al. General synthesis of a diatomic catalyst library via a macrocyclic precursor-mediated approach. J Am Chem Soc, 2023, 145(8): 4819 doi: 10.1021/jacs.2c13886
[73] Wu H L, Huang L Q, Timoshenko J, et al. Selective and energy-efficient electrosynthesis of ethylene from CO2 by tuning the valence of Cu catalysts through aryl diazonium functionalization. Nat Energy, 2024, 9(4): 422 doi: 10.1038/s41560-024-01461-6
[74] Wang B Q, Wang M, Fan Z T, et al. Nanocurvature-induced field effects enable control over the activity of single-atom electrocatalysts. Nat Commun, 2024, 15(1): 1719 doi: 10.1038/s41467-024-46175-1
[75] 张俊, 龙涛, 李广环, 等. 双金属Co/Fe‒N‒C催化剂合成及氧还原性能研究. 功能材料, 2024, 55(8):8143 doi: 10.3969/j.issn.1001-9731.2024.08.019 Zhang J, Long T, Li G H, et al. Synthesis and oxygen reduction performance study of bimetallic Co/Fe‒N‒C catalysts. J Funct Mater, 2024, 55(8): 8143 doi: 10.3969/j.issn.1001-9731.2024.08.019
[76] Lv X S, Wei W, Huang B B, et al. High-throughput screening of synergistic transition metal dual-atom catalysts for efficient nitrogen fixation. Nano Lett, 2021, 21(4): 1871 doi: 10.1021/acs.nanolett.0c05080
[77] Ding T, Liu X K, Tao Z N, et al. Atomically precise dinuclear site active toward electrocatalytic CO2 reduction. J Am Chem Soc, 2021, 143(30): 11317 doi: 10.1021/jacs.1c05754
[78] Zhao X, Zhao K, Liu Y M, et al. Highly efficient electrochemical CO2 reduction on a precise homonuclear diatomic Fe–Fe catalyst, ACS Catalysis, 2022, 12(18): 11412
[79] Pu T C, Ding J Q, Zhang F X, et al. Dual atom catalysts for energy and environmental applications. Angew Chem Int Ed, 2023, 62(40): e202305964 doi: 10.1002/anie.202305964
[80] Song P Y, Zhu P, Su X R, et al. Microenvironment modulation in carbon-supported single-atom catalysts for efficient electrocatalytic CO2 reduction. Chem, 2022, 17(20): e202200716
[81] Xu Y F, Gao W Q, Chen Z X, et al. Cation-dependent impact of CO2 on Cu-catalyzed electrochemical CO reduction reaction. ACS Catal, 2024, 14(14): 10829 doi: 10.1021/acscatal.4c02498
[82] Zhao Q P, Shi W X, Zhang J W, et al. Photo-induced synthesis of heteronuclear dual-atom catalysts. Nat Synth, 2024, 3(4): 497 doi: 10.1038/s44160-024-00486-9
[83] Wang J, Huang Z Q, Liu W, et al. Design of N-coordinated dual-metal sites: A stable and active Pt-free catalyst for acidic oxygen reduction reaction. J Am Chem Soc, 2017, 139(48): 17281 doi: 10.1021/jacs.7b10385
[84] Han X P, Ling X F, Yu D S, et al. Atomically dispersed binary Co‒Ni sites in nitrogen-doped hollow carbon nanocubes for reversible oxygen reduction and evolution. Adv Mater, 2019, 31(49): 1905622 doi: 10.1002/adma.201905622
[85] Liu M, Li N, Cao S F, et al. A “pre-constrained metal twins” strategy to prepare efficient dual-metal-atom catalysts for cooperative oxygen electrocatalysis. Adv Mater, 2022, 34(7): 2107421 doi: 10.1002/adma.202107421
[86] Cheng H Y, Wu X M, Feng M M, et al. Atomically dispersed Ni/Cu dual sites for boosting the CO2 reduction reaction. ACS Catal, 2021, 11(20): 12673 doi: 10.1021/acscatal.1c02319
[87] Chen Z Q, Huang A J, Yu K, et al. Fe1N4–O1 site with axial Fe–O coordination for highly selective CO2 reduction over a wide potential range. Energy Environ Sci, 2021, 14(6): 3430 doi: 10.1039/D1EE00569C
[88] Deng Y C, Zhao J, Wang S F, et al. Operando spectroscopic analysis of axial oxygen-coordinated single-Sn-atom sites for electrochemical CO2 reduction. J Am Chem Soc, 2023, 145(13): 7242 doi: 10.1021/jacs.2c12952
[89] Zhang L J, Jin N, Yang Y B, et al. Advances on axial coordination design of single-atom catalysts for energy electrocatalysis: A review. Nanomicro Lett, 2023, 15(1): 228
[90] Zhu E Z, Sun C H, Shi C Y, et al. Isolated single-atom Fe‒N4O1 catalytic site from a pre-oxidation strategy for efficient oxygen reduction reaction. Chem Eng J, 2023, 463: 142468 doi: 10.1016/j.cej.2023.142468
[91] Zhang Y L, Liu B, Dai Y K, et al. Electronic delocalization regulates the occupancy and energy level of Co3 dz2 orbitals to enhance bifunctional oxygen catalytic activity. Adv Funct Mater, 2022, 32(49): 2209499 doi: 10.1002/adfm.202209499
[92] Li M, Wang M M, Liu D Y, et al. Atomically-dispersed NiN4‒Cl active sites with axial Ni‒Cl coordination for accelerating electrocatalytic hydrogen evolution. J Mater Chem A, 2022, 10(11): 6007 doi: 10.1039/D1TA08287F
[93] Guo Y B, Yao S, Xue Y Y, et al. Nickel single-atom catalysts intrinsically promoted by fast pyrolysis for selective electroreduction of CO2 into CO. Appl Catal B Environ, 2022, 304: 120997 doi: 10.1016/j.apcatb.2021.120997
[94] Yang X, Cheng J, Yang X, et al. Boosting electrochemical CO2 reduction by controlling coordination environment in atomically dispersed Ni@N xC y catalysts. ACS Sustainable Chem Eng, 2021, 9(18): 6438 doi: 10.1021/acssuschemeng.1c01364
[95] Li K, Zhang S B, Zhang X L, et al. Atomic tuning of single-atom Fe‒N‒C catalysts with phosphorus for robust electrochemical CO2 reduction. Nano Lett, 2022, 22(4): 1557 doi: 10.1021/acs.nanolett.1c04382
[96] Shen H J, Gracia-Espino E, Ma J Y, et al. Atomically FeN2 moieties dispersed on mesoporous carbon: A new atomic catalyst for efficient oxygen reduction catalysis. Nano Energy, 2017, 35: 9 doi: 10.1016/j.nanoen.2017.03.027
[97] Zhou W L, Su H, Li Y L, et al. Identification of the evolving dynamics of coordination-unsaturated iron atomic active sites under reaction conditions. ACS Energy Lett, 2021, 6(9): 3359 doi: 10.1021/acsenergylett.1c01316
[98] Liu J J, Gong Z C, Allen C, et al. Edge-hosted Fe‒N3 sites on a multiscale porous carbon framework combining high intrinsic activity with efficient mass transport for oxygen reduction. Chem Catal, 2021, 1(6): 1291 doi: 10.1016/j.checat.2021.09.012
[99] Han G K, Zhang X, Liu W, et al. Substrate strain tunes operando geometric distortion and oxygen reduction activity of CuN2C2 single-atom sites. Nat Commun, 2021, 12(1): 6335 doi: 10.1038/s41467-021-26747-1
[100] Chen W X, Pei J J, He C T, et al. Single tungsten atoms supported on MOF-derived N-doped carbon for robust electrochemical hydrogen evolution. Adv Mater, 2018, 30(30): 1800396 doi: 10.1002/adma.201800396
[101] Liu L Y, Xiao T, Fu H Y, et al. Construction and identification of highly active single-atom Fe1‒NC catalytic site for electrocatalytic nitrate reduction. Appl Catal B Environ, 2023, 323: 122181 doi: 10.1016/j.apcatb.2022.122181
[102] Cheng X F, He J H, Ji H Q, et al. Coordination symmetry breaking of single-atom catalysts for robust and efficient nitrate electroreduction to ammonia. Adv Mater, 2022, 34(36): 2205767 doi: 10.1002/adma.202205767
[103] Zhang L B, Feng J Q, Liu S J, et al. Atomically dispersed Ni‒Cu catalysts for pH-universal CO2 electroreduction. Adv Mater, 2023, 35(13): 2209590 doi: 10.1002/adma.202209590
[104] Yun R R, Zhan F Y, Wang X J, et al. Design of binary Cu–Fe sites coordinated with nitrogen dispersed in the porous carbon for synergistic CO2 electroreduction. Small, 2021, 17(4): 2006951 doi: 10.1002/smll.202006951
[105] Jiao J Q, Yuan Q, Tan M J, et al. Constructing asymmetric double-atomic sites for synergistic catalysis of electrochemical CO2 reduction. Nat Commun, 2023, 14(1): 6164 doi: 10.1038/s41467-023-41863-w
[106] Liu W, Li H Q, Ou P F, et al. Isolated Cu-Sn diatomic sites for enhanced electroreduction of CO2 to CO. Nano Res, 2023, 16(7): 8729 doi: 10.1007/s12274-023-5513-5
[107] Han L L, Ou P F, Liu W, et al. Design of Ru‒Ni diatomic sites for efficient alkaline hydrogen oxidation. Sci Adv, 2022, 8(22): eabm3779 doi: 10.1126/sciadv.abm3779
[108] Guo Z Y, Zhu H W, Yang G, et al. Synergistic engineering of heteronuclear Ni–Ag dual-atom catalysts for high-efficiency CO2 electroreduction with nearly 100% CO selectivity. Chem Eng J, 2023, 476: 146556 doi: 10.1016/j.cej.2023.146556
[109] Yang G G, Zhu J W, Yuan P F, et al. Regulating Fe-spin state by atomically dispersed Mn‒N in Fe‒N‒C catalysts with high oxygen reduction activity. Nat Commun, 2021, 12(1): 1734 doi: 10.1038/s41467-021-21919-5
[110] Tang B, Zhou Y N, Ji Q Q, et al. A Janus dual-atom catalyst for electrocatalytic oxygen reduction and evolution. Nat Synth, 2024, 3(7): 878 doi: 10.1038/s44160-024-00545-1
[111] Xiao M L, Zhu J B, Li S, et al. 3d-orbital occupancy regulated Ir-co atomic pair toward superior bifunctional oxygen electrocatalysis. ACS Catal, 2021, 11(14): 8837 doi: 10.1021/acscatal.1c02165
 下载:
下载:
计量
- 文章访问数: 166
- HTML全文浏览量: 49
- PDF下载量: 29
